Un défaut de réparation de l'ADN
Rédigé par Jean-Yves Dupont
Relu par :
La symptomatique
Xeroderma pigmentosum est une affection héréditaire autosomale récessive (incidence : 1 personne sur 250 000). Elle est responsable d'une extrême sensibilité aux UV solaires. Sans protection, les sujets subissent de sérieux dommages à la peau et aux yeux.
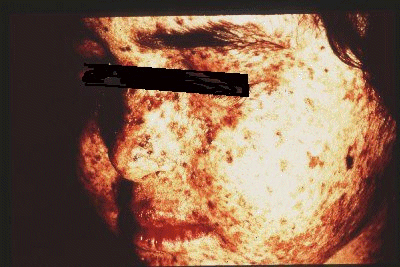 | La plupart des patients développent très tôt de nombreuse taches de rousseur. Des expositions au soleil entraînent des tâches sombres sur la peau, un amincissement de celle-ci, des kératoses et des cancers. Ces modifications font penser à celles que développent les personnes âgées mais elles interviennent dès l'enfance. Source : "An introduction to genetic analysis". Anthony J.F. Griffiths et al. 6th edition, W.H. Freeman and company |
Les yeux sont souvent douloureux, irrités et larmoyants.
20% des sujets présentent des atteintes nerveuses et un retard de développement.
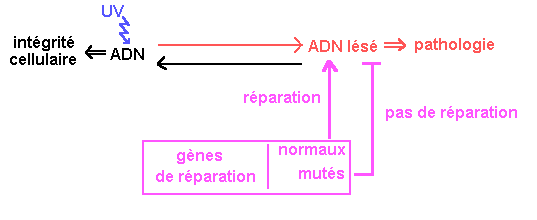
Xeroderma pigmentosum est une affection radio-induite.
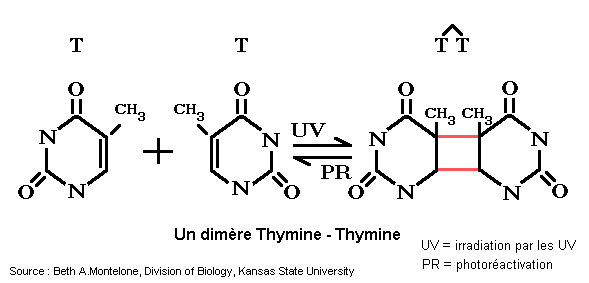
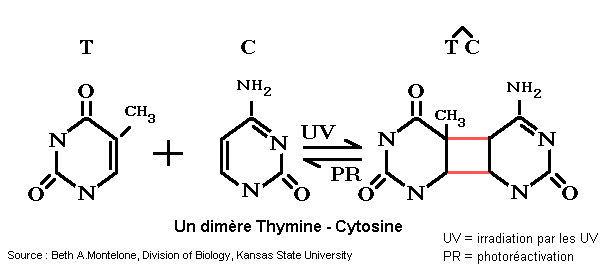
Elle résulte de l'atteinte de l'ADN des cellules (en particulier des cellules cutanées) par les rayons ultra-violets (surtout les UV-B). Les lésions les plus fréquentes sont des lésions "simple brin" comme la formation de diméresT-T ou T-C entre les pyrimidines qui bloquent la transcription et la réplication.
Habituellement, 80% de l'ADN est réparé en 15 minutes. Dans le cas du Xeroderma, le taux de réparation n'est que de 10%.
{kind=link}
{kind=link}
Xeroderma résulte de mutations somatiques qui perdurent par défaut de réparation de l'ADN
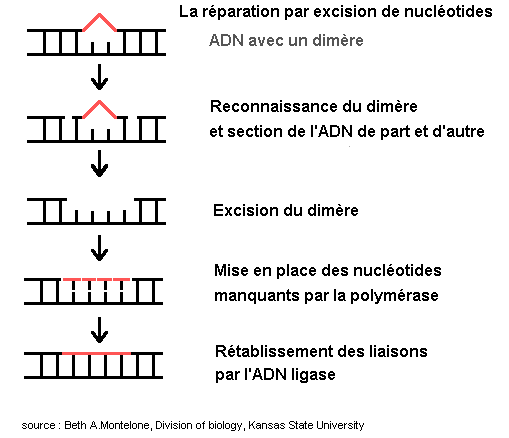
La plus importante voie de réparation est l'excision des nucléotides altérés. Cette opération comporte plusieurs phases : reconnaissance du dimère, excision, remplacement des nucléotides, rétablissement des liaisons.
{kind=link}
La reconnaissance fait intervenir les produits des génes ERCC1, XPA et XPF qui interagissent avec le facteur de transcription TFIIH (produits des gènes de réparation XPB et XPD).
L'excision est réalisée par les produits des gènes ERCC1 et XPG. L'opération est achevée par l'action de la polymérase et de la ligase.
La protéine de réparation résultant de l'expression du gène XPA comporte 273 acides aminés. C'est une protéine en "doigts de Zinc".
Le gène XPA (25 Kb) est localisé sur le chromosome 9 et comporte 6 exons.
Des mutations affectant ces exons entrainent une altération totale ou partielle de la fonction réparatrice.
Quelques mutations retenues
Des substitutions
| Noms des allèles dans la banque de données | Nature et position de la mutation | Conséquences phénotypiques | Incidence sur la protéine |
| xpa_norm | pas d'hypersensibilité aux UV (réparation normale) | ||
| xpa_mut1 | G -> A 216 | pas d'hypersensibilité aux UV (réparation normale) | aucune |
| xpa_mut2 | G -> C 381 | faible sensibilité (légère altération de la fonction réparatrice) | Asp->His 127 |
| xpa_mut3 | C -> T 457 | forte sensibilité aux UV (pas de fonction réparatrice) | protéine écourtée |
| xpa_mut4 | A -> G 557 | sensibilité intermédiaire (diminution de la fonction réparatrice) | His -> Arg 186 |
| xpa_mut5 | T -> A 174 | forte sensibilité aux UV (pas de fonction réparatrice) | protéine écourtée |
| xpa_mut6 | C -> T 508 | forte sensibilité aux UV (pas de fonction réparatrice) | protéine écourtée |
| xpa_mut7 | T -> G 139 T -> A 148 T -> G 202 T -> A 211 | très forte sensibilité (pas de fonction réparatrice) | Cys -> Gly 47 Cys -> Ser 50 Cys -> Gly 68 Cys -> Ser 71 |
Des délétions
| Noms des allèles dans la banque de données | Nature et importance de la mutation | Conséquences phénotypiques | Incidence sur la protéine |
| xpa_mut8 | delétion de A 59 à A 79 | forte sensibilité aux UV (pas de fonction réparatrice) | séquence modifiée protéine écourtée |
| xpa_mut9 | délétion C 200 | forte sensibilité aux UV (pas de fonction réparatrice) | séquence modifiée protéine écourtée |
| xpa_mu10 | délétion de 172 à 177 (TATCT) | forte sensibilité aux UV (pas de fonction réparatrice) | séquence modifiée protéine écourtée |
Conclusion