
Origine et évolution du Sars-CoV-2 responsable de la pandémie Covid-19
1. Objectifs de la recherche
La recherche sur l’origine du Sars-CoV-1 a abouti à l’idée que c’est un coronavirus hébergé par une ou plusieurs espèces de Chauve-souris qui était à l’origine du coronavirus humain (chauve-souris réservoirs de virus). Toutefois la transmission du coronavirus à l’Homme n’a pas été directe mais a impliqué au moins une espèce intermédiaire, la Civette.
Dès l’élucidation du génome de Sars-CoV-2 au tout début de l’année 2020, les chercheurs ont voulu savoir si des coronavirus de Chauve-souris pouvaient en être à l’origine et si la transmission à l’Homme s’était réalisée grâce à un hôte intermédiaire (l’équivalent de la Civette pour le Sars-CoV-1).
La méthodologie de la recherche est semblable à celle utilisée pour le Sars-CoV-1 et repose sur la comparaison des séquences de génomes de coronavirus trouvés chez divers animaux (en premier lieu les Chauve-souris) avec celle du Sars-CoV-2 . Elle se prolonge par la réalisation d’arbres phylogénétiques traduisant les relations de parenté entre les divers génomes séquencés.
2. Un coronavirus de Chauve-souris à l’origine du Sars-Cov2 ?
Pour aborder ce sujet, on dispose des documents suivants :
Fichier pour Anagène : Fichier Sars-divers bat et pangolin.edi
Arbre phylogénétique de nombreux coronavirus (Extrait du site Nexstrain : Phylogeny of sars-like betacoronavirus including novel coronavirus Sars-CoV-2)
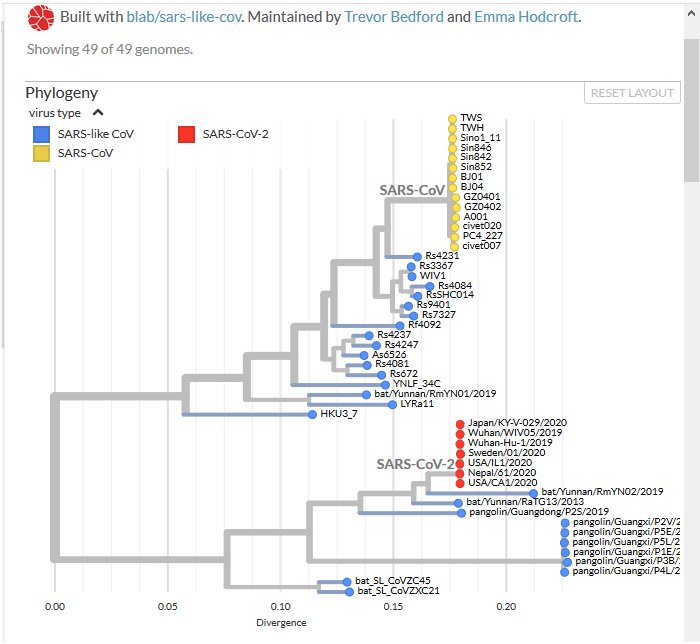
En jaune : génomes de Sars-CoV-1 ; en rouge :génomes de Sars-CoV-2 ; en bleu : génomes de coronavirus de Chauve-souris et de Pangolins.
Outre la séquence du Sars-CoV-2, le fichier pour Anagène comprend celles de 3 génomes de coronavirus trouvés chez des Chauves-souris (Sars BatRaTG13, Sars BatZXC21 et Sars BatZC45) ainsi que celle trouvée chez un Pangolin. Les chercheurs au courant d’informations indiquant que des Pangolins manifestaient des symptômes proches de ceux de la Covid-19 humaine, ont trouvé des coronavirus chez ces animaux malades et ont séquencé leurs génomes.
Exploitation pédagogique de ces informations
La comparaison avec Anagène des séquences des génomes de coronavirus animaux avec celle du Sars-CoV-2 indique une forte similitude, supérieure à 85%. C’est le coronavirus RaTG13 trouvé chez une Chauve-souris qui a la plus forte similitude, légèrement supérieure à 96%. Celle des deux autres coronavirus de Chauve-souris est environ de 87% et celle du coronavirus de Pangolin de 90%.
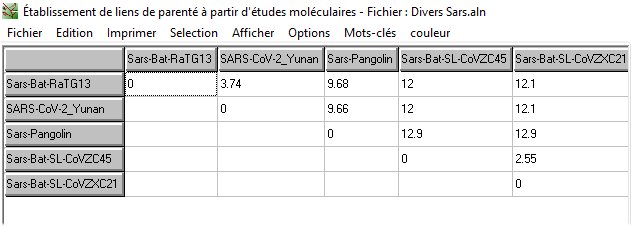
Matrice des différences exprimées en pourcentages obtenue avec Phylogène.
A partir des informations sur la similitude des séquences des coronavirus, on peut construire un arbre phylogénétique (éventuellement avec l’aide de phylogène).
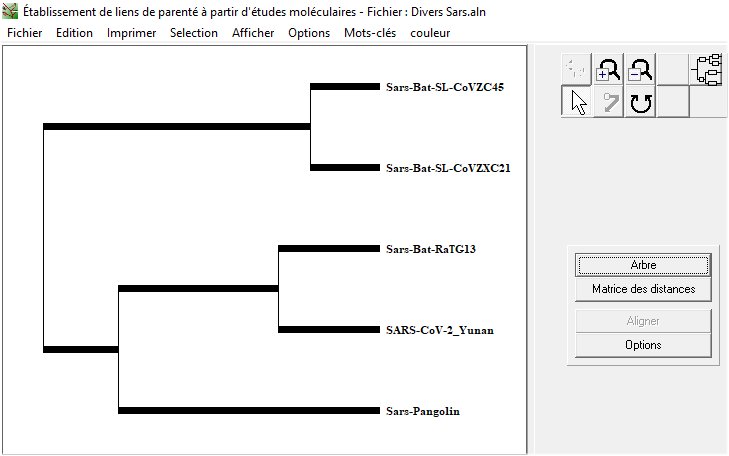
L’arbre phylogénétique de Nexstrain traduit l’histoire évolutive des deux virus émergents apparus en Chine, celle du Sars-CoV-1 (autrefois on disait Sars-CoV), de 2002-2003 et celle du Sars-CoV-2 de 2019-2020. Les deux arbres indiquent que ces deux virus humains ont pour origine des coronavirus de Chauve-souris (espèce réservoir). Mais l’arbre indique aussi deux histoires évolutives différentes liées au fait que ce sont des coronavirus différents de Chauve-souris qui sont à l’origine des deux coronavirus humains. Cela est en relation avec le fait que les Chauve- souris de Chine hébergent une grande variété de coronavirus. Un même animal peut d’ailleurs héberger plus d’une souche virale. Cela rend possible une évolution vers un autre coronavirus humain dans l’avenir.
L’arbre de Nexstrain confirme aussi que parmi tous les coronavirus séquencés le coronavirus de RaTG13 est le plus proche du Sars-CoV-2.
3. Une transmission directe de la Chauve-souris à l’Homme ?
La similitude très forte de 96% du génome du coronavirus de RaTG13 avec celui du Sars-CoV-2 peut laisser penser que le coronavirus humain peut provenir d’un coronavirus proche de RaTG13 après une évolution l’ayant rendu capable de franchir la barrière d’espèce, suivie d’une évolution dans l’espèce humaine.
Pour tester cette hypothèse, on dispose des documents suivants :
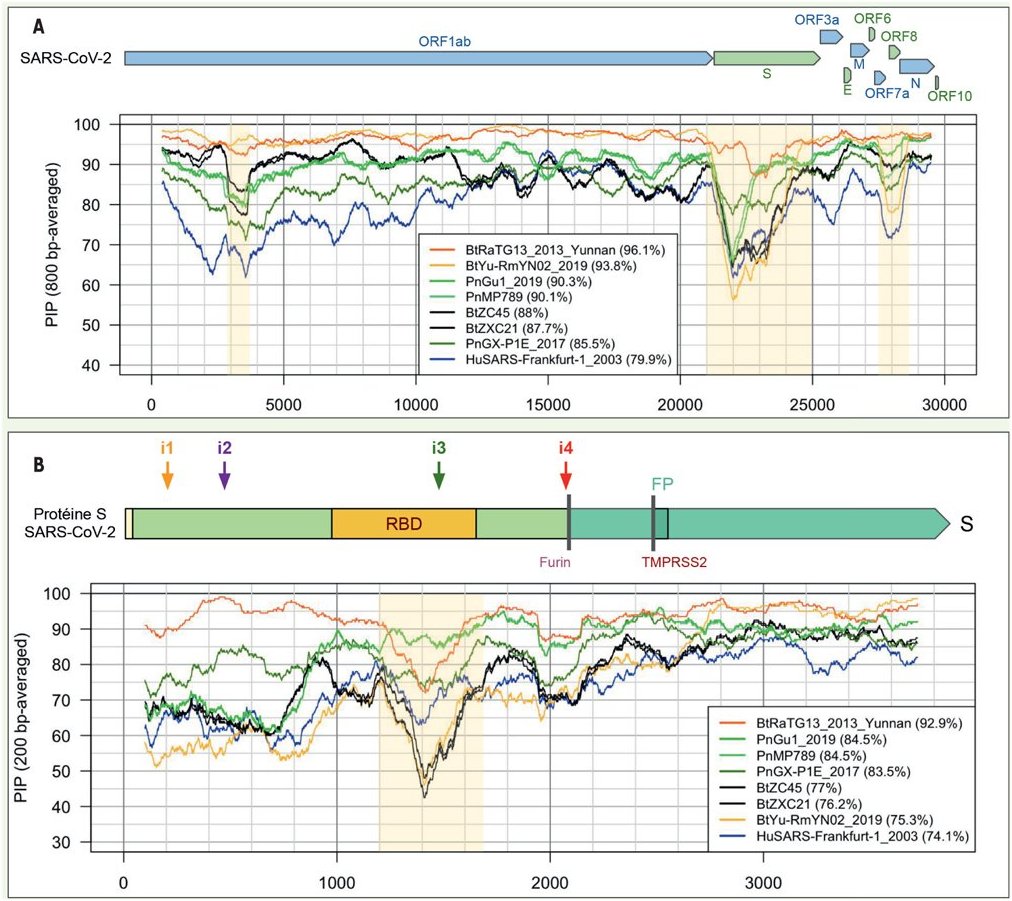
Image 2A : Le taux d’identité entre SARS-CoV-2 et les autres coronavirus varie selon la position dans le génome. Le niveau 100 % correspond au génome de référence du SARS-CoV-2. A. Pourcentage d’identité entre SARS-CoV-2 et d’autres coronavirus le long du génome entier (fenêtres glissantes de 800 nucléotides).
Erwan Sallard et al. Retrouver les origines du SARS-CoV-2 dans les phylogénies de coronavirus. Med Sci (Paris). Volume 36, Number 8-9, Août–Septembre 2020.
- Fichier pour Anagène des séquences protéiques des RBD de Sars-CoV-2 et de RaTG13.
Spike-CoV2-CoV1-Publication Morse.edi
Exploitation pédagogique
Il y a environ 4% de différence entre le génome complet du Sars-Cov-2 et celui de RaTG13 de Chauve-souris. Les données sur le pourcentage d’identité des deux génomes tout au long de leurs séquences permet d’affiner cette estimation globale. Les figures prennent en compte les séquences de plusieurs coronavirus mais il suffit de considérer la séquence du RaTG13 aussi bien pour le graphique relatif au génome global que pour celui relatif au gène codant pour la seule protéine S. Pour interpréter ces graphiques, il faut bien voir que c’est le pourcentage d’identité entre le génome du coronavirus considéré et celui du Sars-Cov-2 qui est visualisé. Le génome du sars -Cov-2 sert de référence et correspond donc à une identité de100%.
On constate que la séquence de RaTG13 a partout un pourcentage de similitude nettement supérieur à 90% sauf entre les nucléotides 21.000 et 24.500 correspondant à la région du génome codant pour la protéine S où le pourcentage tombe sous 90%.
La figure indique que cette région code pour le RBD c’est-à-dire le domaine de liaison de la protéine S (spike) au récepteur ACE2 des cellules humaines infectables.
Le fichiers de séquences protéiques des RBD des protéines S de Sars-Cov-2 et de RaTG13 confirment et précisent Les informations extraites des graphiques. La comparaison avec Anagène des séquences des RBD des deux virus indique que le pourcentage d’identité entre les deux séquences est inférieure à 90% donc nettement inférieure à l’identité entre les génomes complets.
Conclusion : La protéine S (Spike) joue un rôle crucial dans la capacité du virus à pénétrer dans les cellules humaines (de l’épithélium pulmonaire par exemple) et donc à débuter un cycle infectieux. Les différences importantes entre les RBD des protéines S du coronavirus RaTG13 de Chauve-souris et du Sars-CoV-2 humain laissent à penser que le coronavirus RaTG13 de Chauve-souris doit avoir une efficacité faible pour parasiter les cellules humaines. Cela réfute l’hypothèse que le RaTG13 ait pu être à l’origine du Sars-CoV-2 humain suite à une transmission directe de la Chauve-souris à l’Homme.
4. Le Pangolin et la question de son implication dans l’histoire évolutive du Sars-CoV-2
Pour aborder ce sujet, on dispose de fichiers de séquences déjà fournis pour les points précédents :
- Le fichier Sars-divers bat et pangolin edi. (fourni pour le point 2) qui permet de comparer la séquence du génome complet du coronavirus du pangolin à celle des génomes de Sars-Cov-2 et RaTG13
- Le fichier des séquences protéiques des RBD des protéines S de Sars-Cov-2, RaTG13 et du Pangolin (fichier déjà fourni pour le point 3) qui permet de dégager les caractéristiques du RBD du Pangolin par rapport aux deux autres.
On dispose aussi de documents complémentaires déjà fournis pour envisager les points précédents :
- arbre phylogénétique des coronavirus établi à partir des séquences des génomes complets permettant de préciser avec quels virus, celui du Pangolin est le plus proche (arbre fourni pour le point 2).
- Pourcentage d’identité tout au long de la séquence du gène S des coronavirus RaTG13 et du pangolin avec le Sars-Cov-2 (Figures déjà fournies pour traiter le point 2)
- Enfin comme document propre à ce quatrième point on dispose des arbres phylogénétiques construits à partir de la comparaison des séquences du gène S de plusieurs coronavirus. (Origine : Nexstrain).
- Domaine de fixation au récepteur ACE.edi
- Domaine de fixation au récepteur ACE.aln
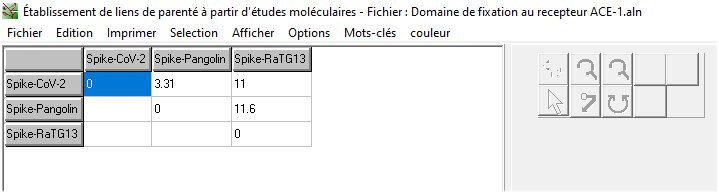
Matrice des différences exprimées en pourcentages obtenue avec Phylogène.
Exploitation pédagogique
- Informations fournies par la comparaison des génomes complets de coronavirus
Le génome du coronavirus d’un Pangolin a une séquence présentant 90% d’identité avec celle du Sars-CoV-2 ce qui est nettement inférieur au pourcentage d’identité du coronavirus RaTG13 de la Chauve-souris (96,2%). Cette information suggère qu’il est très peu probable que le Pangolin ait été l’hôte intermédiaire impliqué dans la transmission du Sars-CoV-2 à l’Homme. Dans le cas du Sars-CoV-1, le coronavirus de Civette avait une identité proche de 100% avec celui-ci, ce qui indiquait clairement que le coronavirus humain provenait de la transmission du coronavirus de la Civette à l’Homme. La similitude de 90% est insuffisante pour qu’on puisse considérer le Pangolin comme l’hôte intermédiaire ayant transmis le Sars-CoV-2 à l’Homme.
L’arbre de Nexstrain confirme que les coronavirus du Pangolin sont apparentés au Sars-CoV-2 et non au Sars-CoV-1. Il indique aussi que le coronavirus de Guangdong dont la séquence a été fournie dans le fichier de séquences précédent est plus étroitement apparenté avec le Sars-CoV-2 que les autres coronavirus de Pangolin. Cela confirme qu’il est peu probable qu’un coronavirus de Pangolin soit directement à l’origine du Sars-CoV-2.
- Informations fournies par la comparaison des gènes S et plus particulièrement des régions qui codent pour le RBD de la protéine S
Comme vu précédemment, le profil d’identité indique que la séquence du gène S du coronavirus RaTG13 présente une nette baisse de sa similitude avec la séquence de Sars-CoV-2 au niveau d’une région qui code pour le domaine RBD de liaison au récepteur cellulaire ACE2 (RBD) de la protéine S. Or cela n’est pas vrai pour la séquence du coronavirus du Pangolin qui est plus proche de celle de Sars-CoV-2 que ne l’est la séquence du RaTG13.
Dans le RBD du Sars-CoV-2, 6 acides aminés jouent un rôle crucial dans la liaison au récepteur ACE2 des cellules humaines : L120, F151, Q158, S159, N166 et Y170. (L :leucine, F : phénylalanine ; Q : glutamine, S : sérine, N : asparagine, Y tyrosine).
La comparaison des séquences protéiques des RBD des trois coronavirus Sars-CoV-2, RaTG13 et de Pangolin.Cov permet de préciser ce constat.
- Ces séquences ont 181 acides aminés et le tableau traduit les pourcentages d’identité. La séquence du RBD du coronavirus du Pangolin a un pourcentage d’identité de 96,7% avec celle du RBD du Sars-CoV-2 alors que pour le RBD du coronavirus de Chauve-souris, il n’est que de 89%.
- Surtout, on constate que la séquence du RBD du coronavirus du Pangolin possède les mêmes acides aminés que le RBD du Sars-CoV-2 cruciaux pour la liaison avec le récepteur ACE2 et donc l’entrée dans les cellules humaines : L120 ; F151 ; Q158 ; S159 ; N166 et Y170. En revanche à ces sites, le RBD du RaTG13 n’a qu’un seul acide aminé en commun avec le RBD de Sars-CoV-2 : L120. Cela signifie que, par les caractéristiques du RBD de sa protéine S, le coronavirus du Pangolin est plus apte à pénétrer et parasiter les cellules humaines que ne l’est le coronavirus RaTG13 de Chauve-souris.
En résumé : La forte identité globale de la séquence du génome de RaTG13 avec celle du Sars-CoV-2 suggère qu’un coronavirus de Chauve-Souris peut être à l’origine du Sars-CoV-2. Mais la dissemblance dans les séquences du gène S (et en particulier de la région qui code pour le RBD), indiquent qu’il n’y a pas eu de transmission directe d’un coronavirus de la Chauve-souris à l’Homme.
Les caractéristiques du gène S et surtout de la région codant pour le RBD de la protéine S du coronavirus du Pangolin, montrent qu’un tel virus est susceptible de pénétrer dans les cellules humaines munies du récepteur ACE2. Cependant l’identité globale des génomes du coronavirus du Pangolin avec celui du Sars-CoV-2 est trop insuffisante pour qu’on puisse affirmer que le Pangolin est un hôte intermédiaire qui a transmis le Sars-Cov2 à l’Homme . L’origine du Sars-CoV-2 n’a pas encore été résolue.
5 . La recombinaison génétique, un mécanisme important de l’évolution des coronavirus.
Ainsi le génome de Sars-CoV-2 comprend à une grande partie de sa séquence avec une forte identité à celle du coronavirus de Chauve-souris, dans laquelle est insérée, dans la région codant pour le gène S, une séquence très fortement identique à celle d’un coronavirus de Pangolin. Cela suggère que dans l’histoire évolutive de Sars-CoV-2, il y a eu un mécanisme de recombinaison génétique associant un fragment d’un coronavirus de Chauve-Souris à un fragment du génome d’un coronavirus de Pangolin.
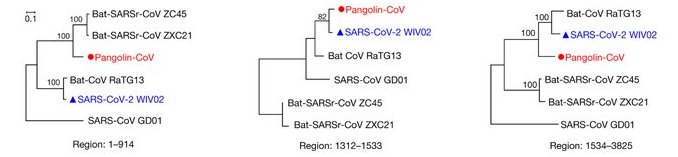
Cette possibilité de recombinaison génétique chez les coronavirus est aussi mise en évidence par les arbres phylogénétiques établis à partir de la comparaison des séquences de diverses régions des génomes de plusieurs coronavirus. Ainsi la figure ci-dessous montre les arbres phylogénétiques résultant de la comparaison du gène S de plusieurs coronavirus : 3 coronavirus de Chauve-souris RaTG13, ZCX21, ZC45, un coronavirus de Pangolin et deux coronavirus humains : Sars-Cov-2 et Sars-Cov61 (Sars-Cov GD01). (Origine : Nexstrain).
Dans la région allant du premier au 914ème nucléotide du gène S, le génome du coronavirus du Pangolin est plus apparenté aux génomes des coronavirus ZXC21 et ZC45 de Chauve-souris, alors que pour les régions 1312-1533 et 1534,-3825, il est davantage proche de RaTG13 de Chauve-souris et du Sars-CoV-2.
Dans la région 1312-1533 Le coronavirus du pangolin est davantage apparenté avec le Sars-CoV-2 que ne l’est le RaTG13 alors que c’est le contraire pour les autres régions.
Ces contradictions dans les phylogénies établies à partir de différentes régions des séquences des génomes de coronavirus sont la traduction d’intervention de recombinaisons génétiques dans l’histoire évolutive des coronavirus.
On a pu préciser chez les coronavirus de Chauve-souris comment se réalise cette recombinaison génétique.
Les chercheurs chinois ont mis en évidence que les populations de Chauve-souris du sud de la Chine hébergent une grande diversité de coronavirus. En outre un même animal peut héberger des coronavirus différents et, plus précisément, une cellule peut être parasitée par deux coronavirus appartenant à deux souches différentes. Dans une telle cellule, l’ARN polymérase qui entame la réplication du génome d’un virus peut sauter sur le brin d’ARN de l’autre virus et le répliquer avant d’achever la réplication du premier virus. On obtient ainsi un génome chimérique d’un autre coronavirus.
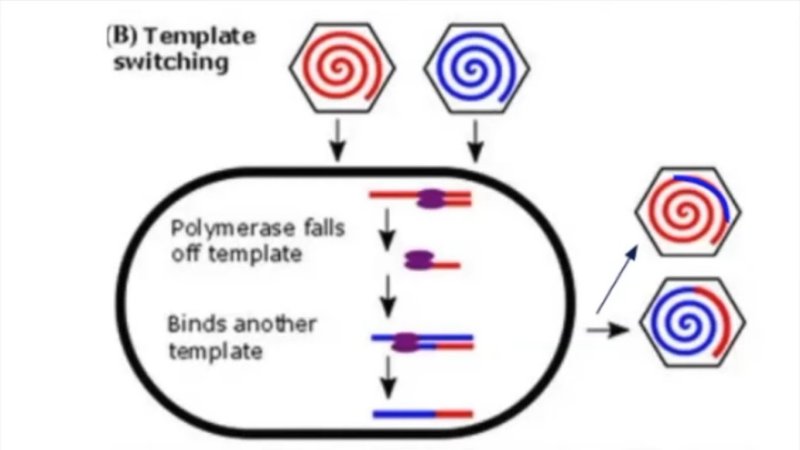
D'après : Covid-19 : approches scientifiques et enjeux sociétaux #1. Virologie moléculaire (Etienne Decroly). Université de Marseille. Youtube. Durée 1h56 minutes et 29 secondes.
Légende : en rouge le génome d’un coronavirus ; en bleu le génome d’un autre coronavirus. Ces deux coronavirus parasitent la même cellule. Le schéma évoque la réplication de ces deux génomes par la polymérase.
En guise de conclusion.
- L’apparition de coronavirus émergents comme le Sars-CoV-1, le MERS et le Sars-CoV-2 traduit l’inéluctable évolution des génomes des virus comme cela est le cas des génomes des organismes procaryotes et eucaryotes.
- Les deux modalités de l’évolution des génomes des coronavirus sont les mutations plus ou moins ponctuelles et les recombinaisons génétiques entre génomes de coronavirus différents. Ces recombinaisons aboutissant à des génomes chimériques ont lieu dans les cellules parasitées lors de la réplication de l’ARN de deux génomes viraux plus ou moins différents. La recombinaison génétique est un mécanisme qui peut engendrer une évolution rapide des génomes et favoriser l’apparition de virus susceptibles de parasiter de nouvelles espèces.
- Les mutations sont des phénomènes ayant lieu constamment comme le montre l’extrême diversité des génomes de Sars-CoV-2 un an après les premiers génomes séquencés. L’apparition de nouveaux mutants est favorisée par la reproduction très rapide des coronavirus.
- Ces innovations génétiques par mutations et recombinaisons génétiques donnent prise à la sélection naturelle (voir le paragraphe sur le Sars-CoV-1). Beaucoup de mutations sont neutres car n’ayant pas d’effet sur la capacité du virus à parasiter les cellules humaines et à se multiplier. Cela semble être le cas des mutations apparues lors de la première année d’évolution du Sars-CoV-2 chez l’Homme.
- La diversité des coronavirus de Chauve-Souris notamment en Chine, rend possible l’apparition de nouveaux virus émergents dans l’avenir.
- Certaines espèces, comme les Visons, ont un récepteur ACE2 très proche de celui l’Homme. Cela rend possible la transmission du Sars-CoV-2 humain aux visons. Inéluctablement, chez les Visons, il peut évoluer. Certains mutants en se transmettant à l’homme peuvent avoir une virulence plus grande. Cela pose le problème du danger des élevages d’animaux sauvages et du risque sanitaire qu’ils peuvent engendrer.