
DMD - Exploitation pédagogique
DMD - Exploitation pédagogique
Jean-Claude Hervé. Ex IA-IPR SVT. Académie de Versailles.
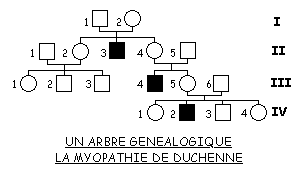
1. Informations fournies par un arbre généalogique
2. Les caractéristiques du gène DMD et de la dystrophine
3. Retour à l’arbre généalogique précédent : l’apport du diagnostic moléculaire
4. Origine de la myopathie dans une famille où un seul enfant est atteint
6. Cas d’une famille où une fille est atteinte de la myopathie de Duchenne
|
L'exploitation des données est illustrée avec le logiciel Anagène. Il est possible d'utiliser Géniegen (Jean-François Madre) et Géniegen2 (Philippe Cosentino) qui ouvrent également les fichiers de séquences au format d'Anagène. Géniegen2 propose une version qui fonctionne en ligne et une autre, à télécharger, pour une utilisation hors ligne. Attention : Les séquences du gène de la dystrophine étant assez longues, il est conseillé de limiter le nombre de séquences à comparer simultanément pour ne pas dépasser les capacités de traitement de ces logiciels. |
1. Informations fournies par un arbre généalogique
Les raisonnements vus en classe de première sont réinvestis pour l’analyse de l’arbre fourni, en particulier pour ce qui concerne la dominance ou la récessivité des allèles. Ainsi les élèves doivent aboutir à la conclusion que les allèles à l’origine de la myopathie de Duchenne sont récessifs puisque l’arbre montre trois cas de garçons myopathes dont les parents ne sont pas atteints. A noter qu’après avoir discuté de la localisation chromosomique du gène DMD, on pourra faire réfléchir sur le fait que la notion de récessivité ne s’applique en réalité qu’au cas des femmes hétérozygotes puisque les garçons ne possèdent qu’un seul allèle du gène DMD.
{kind=link}
La recherche de la localisation chromosomique du gène est propre à la classe de terminale. Pour cela l’élève doit envisager les situations possibles. Il doit ainsi percevoir que la localisation du gène DMD sur un autosome implique que les trois hommes I1, II5 et III6 soient hétérozygotes. Par contre la localisation sur une région propre au chromosome X implique seulement que la femme I2 le soit. Etant donné la fréquence très faible des allèles cause de la myopathie, la localisation autosomale est la moins probable. C’est l’analyse d’autres arbres généalogiques aboutissant aux mêmes observations qui a permis de généraliser par la seule analyse d’arbres, la localisation sur le chromosome X.
L’hypothèse de la localisation du gène DMD sur la région propre au chromosome Y est réfutée par le fait que dans ce cas un homme non myopathe ne peut avoir un fils myopathe, contrairement à ce que montre l’arbre.
Le couple formé par la femme III1 et son conjoint non myopathe ne peut avoir une fille myopathe car le père transmet à sa fille le chromosome X porteur d’un allèle « normal » dominant. La probabilité que le couple ait un garçon myopathe dépend du génotype de la femme III1 : il faut qu’elle soit hétérozygote Xm+ Xm. Il y a une probabilité de 1/2 pour que la femme I2 nécessairement hétérozygote (elle a un fils myopathe), ait transmis le chromosome Xm à sa fille II2. Si celle-ci est hétérozygote il y a de même une probabilité de 1/2 pour que sa fille III1 le soit aussi. Enfin, si cette dernière l’est il y a aussi 1/2 pour qu’elle transmette à son fils le chromosome Xm. Les conditions étant indépendantes, la probabilité que le couple ait un garçon myopathe est égale au produit des probabilités de chacune des conditions nécessaires à la naissance d’un garçon myopathe soit : 1/2 x 1/2 x 1/2 = 1/8. Et si on considère la naissance d’un enfant myopathe, elle est de 1/8 x 1/2 =1/16.
Ce raisonnement représente l’acquis de la terminale.
2. Les caractéristiques du gène DMD et de la dystrophine
Fichier Gène DMD.edi
C’est avant tout une source d’informations utiles pour la compréhension et l’utilisation des données moléculaires se rapportant aux séquences des divers allèles dans les familles étudiées par la suite.
C’est aussi un support pour une révision des modalités de l’expression d’un gène d’eucaryote comportant exons et introns. Les notions d’ARN pré-messager et d’ARN messager sont citées comme fondamentales dans le programme de première. Implicitement, celles d’exons, d’introns et d’épissage au cours de la maturation de l’ARN pré-messager en ARN messager le sont aussi.
Le fichier Gène DMD.edi comprend 5 séquences : CDS gène DMD ; ARNm gène DMD, début CDS DMD ; début pré-ARNm DMD ; début ARNm DMD.
La comparaison des séquences CDS gène DMD et de l’ARNm montre qu’elles sont identiques sauf que les nucléotides à thymine du CDS sont remplacés par des nucléotides à uracile dans l’ARN messager. C’est donc un support pour une révision simple de la transcription d’un gène.
La comparaison des 3 autres séquences a pour objectif une révision plus complète de la transcription d’un gène eucaryote. La séquence Début gène DMD est formée par les séquences des 10 premiers exons et introns. La longueur des introns du gène DMD est considérable ce qui fait qu’en les prenant in extenso ce n’est pas gérable par les logiciels utilisés au lycée. Aussi avons-nous limité les séquences des introns à 100 nucléotides, ceux signalés dans la banque UMD-DMD.
La comparaison des séquences « début gène DMD » et « début pré-ARNm révèle qu’elles sont identiques (sauf bien sûr le remplacement des T par des U.) ce qui permet de caractériser la première étape de la transcription.
La comparaison des séquences « début pré-ARNm » et « début ARNm » révèle que des régions de la séquence de l’ARN pré-messager ne se retrouvent pas dans l’ARN messager ce qui permet de réviser les notions d’épissage d’exons et d’introns.
L’utilisation du Dotplot permet de traduite graphiquement ces notions.
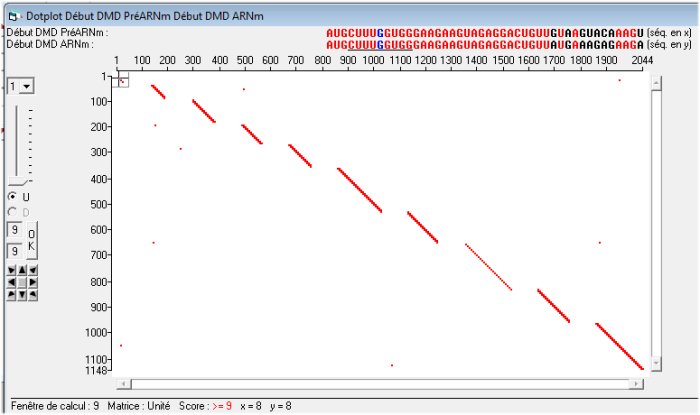
La comparaison avec les logiciels permet de connaître les séquences des 10 premiers exons et introns qu’on peut retrouver dans les informations de la banque.
Si on veut une révision complète de l’expression d’un gène, on peut demander aux élèves d’utiliser la fonction de traduction des logiciels pour traduite l’ARNm.
Remarque
Une simplification a été faite en ce qui concerne les séquences du gène de l’ARN pré-messager et de l’ARN messager. La séquence de l’exon 1 comprend 239 nucléotides mais seulement les 31 derniers seront finalement traduits et font donc partie de la séquence codante du gène, c’est-à-dire du CDS. Cette séquence de 31 nucléotides commence par le triplet ATG.
Lors de la transcription, tout l’exon 1 est transcrit et l’ARN pré-messager commence donc par les 239 nucléotides. Cette séquence se retrouve donc aussi à l’extrémité amont (5') de l’ARNm. Mais l’ARNm n’est pas traduit dans sa totalité. La traduction commence au premier AUG (correspondant au premier ATG de l’exon 1).
Les CDS des allèles des gènes commencent toujours par le triplet ATG. Pour ne pas entrer dans cette discussion, nous avons fait commencer les séquences de l’ARN pré messager et de l’ARN messager par le codon d’initiation de la traduction AUG.
La capacité de l’élève à identifier la séquence des premiers introns est la preuve qu’il maîtrise ces notions. La banque de données des mutations du gène DMD fournit la séquence complète de l’ADNc du gène et celle des exons et introns.
- Les données sur la dystrophine (Séquence de 3685 aa du fichier et schémas de fonctionnement) ne visent pas à une étude extensive de cette protéine musculaire. L’important est que l’élève saisisse qu’elle est indispensable à la stabilité de la membrane des fibres musculaires et repère que son extrémité amino-terminale codée par les premiers exons se lie aux filaments d’actine, et la dernière partie de sa séquence se lie par les derniers exons à un complexe de protéines membranaires. Il est fait appel à ces données plus loin.
3. Retour à l’arbre généalogique précédent : l’apport du diagnostic moléculaire
Le gène DMD a été identifié en 1986. Les techniques récentes de séquençage permettent de connaître les allèles du gène possédés par les personnes d’une famille où 1 ou plusieurs garçons myopathes ont été diagnostiqués.

Toutes les séquences sont des CDS des allèles possédés par les personnes de la famille précédemment envisagée (DMD-Généalogie-Famille1.edi). La simple confrontation entre les données sur le génotype des individus et l’arbre généalogique montre que toutes les femmes possèdent deux allèles du gène alors que tous les hommes n’en possèdent qu’un seul. Cela confirme la localisation tirée précédemment de l’analyse de l’arbre généalogique à savoir que le gène est localisé sur la région propre au chromosome X.

C’est chez le garçon myopathe IV2 qu’a été réalisée la première analyse génétique afin d’identifier la nature de la mutation à l’origine de la myopathie dont il est victime. Comme il ne possède qu’un seul allèle du gène, ce dernier est nécessairement muté. Pour rechercher les caractéristiques de la mutation, on compare la séquence de l’allèle muté possédé par le garçon avec celle de l’allèle de référence.
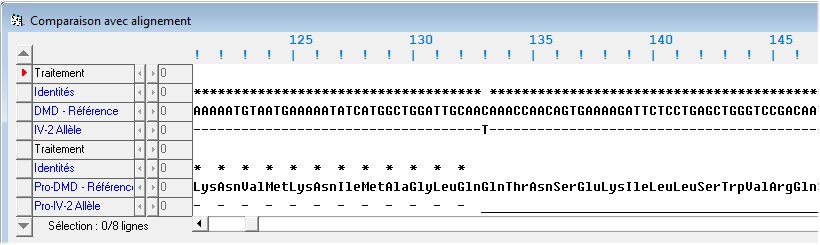
On constate que la mutation est ponctuelle et consiste en la substitution d’un nucléotide à guanine par un nucléotide à thymine. En conséquence le codon CAA133 de l’allèle de référence devient le codon TAA133 dans l’allèle muté. Or le codon TAA est un codon stop. On peut donc s’attendre à ce que la dystrophine codée par l’allèle muté soit raccourcie par rapport à l’allèle « normal ».
Cela peut être vérifié en traduisant l’allèle muté. Dans cette illustration fournie par Anagène, les astérisques et les tirets indiquent des identités entre les deux allèles comparés, et un trait continu une absence de séquence. On constate que la dystrophine mutée ne comprend que 132 acides aminés.
On peut essayer de situer le codon 133 dans la séquence complète du gène pour déterminer l’exon impliqué. On peut utiliser le tableau sur la structure du gène où sont indiquées les longueurs des exons exprimées en nucléotides. Puisque les séquences sont des CDS, la longueur du premier exon à considérer n’est pas de 239 bp mais seulement de 31 bp. Cela oriente vers l’exon 6. La figure ci-dessous fournit la séquence de l’exon 6 fournit par la banque et localise bien la mutation dans cet exon.
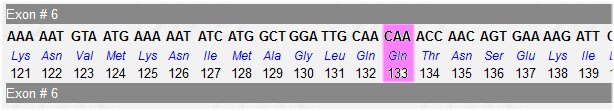
Comme le gène comporte 79 exons, on voit que la dystrophine mutée est considérablement plus courte que la protéine « normale » ce qui l’empêche de jouer son rôle et donc entraîne peu à peu la dégénérescence des fibres musculaires. La myopathie est donc sévère du type myopathie de Duchenne. Sa description complète dans la banque UMD-DMD est la suivante.
Connaissant la nature de la mutation responsable de la myopathie du garçon IV2, le généticien a proposé de déterminer le génotype de plusieurs membres de la famille. La comparaison des allèles possédés par les individus diagnostiqués avec l’allèle de référence est illustrée par le tableau ci-dessous.
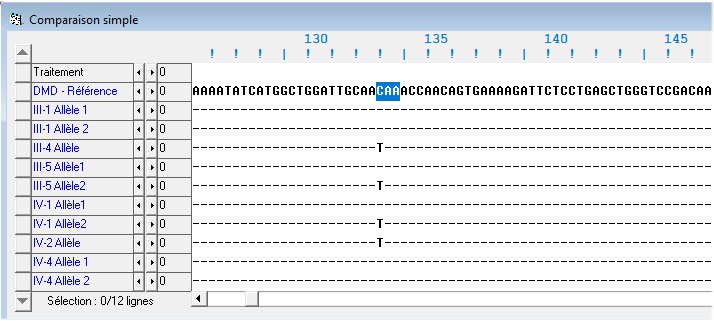
On constate que les mères des garçons myopathes sont toutes hétérozygotes alors que leurs pères ne possèdent qu’un seul allèle « normal ». Cela confirme d’une part que les allèles à l’origine de la myopathie sont récessifs et que d’autre part, l’allèle à l’origine de la myopathie est transmis de génération en génération par l’intermédiaire des femmes.
Enfin, ces informations sur les génotypes des membres de la famille permettent d’évaluer plus précisément le risque que peut avoir un couple d’avoir un enfant myopathe. La femme III1 possède deux allèles « normaux » du gène DMD ce qui indique que le risque d’avoir un enfant myopathe est nul (à moins d’une néo-mutation). Si elle avait été hétérozygote, elle aurait eu 1/2 probabilité d’avoir un garçon myopathe alors que la seule analyse de l’arbre généalogique évaluait cette probabilité à 1/8.
4. Origine de la myopathie dans une famille où un seul enfant est atteint
Cette partie a pour but de répondre à cette phrase des capacités et attitudes du programme : « Recenser et comparer des séquences d’ADN sur des trios père/mère/enfant permettant d’analyser la présence de mutations nouvelles ». Elle suppose qu’on ait envisagé une activité du type de la précédente et donc établi que le gène DMD est situé sur la région propre au chromosome X et avoir acquis les notions de base sur la structure du gène DMD.
Un deuxième objectif en relation avec la structure du gène est la découverte d’un autre type de mutation du gène DMD et de ses conséquences sur le phénotype.
C’est chez le garçon myopathe III4 qu’a été réalisée la première analyse génétique afin d’identifier la nature de la mutation à l’origine de la myopathie dont il est victime. Comme il ne possède qu’un seul allèle du gène DMD, ce dernier est nécessairement muté. Pour rechercher les caractéristiques de la mutation, on compare la séquence de l’allèle muté avec celle de référence.
Ces séquences se trouvent dans le fichier DMD-Généalogie-Famille2.edi
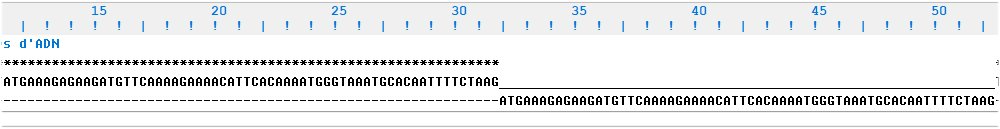
L’échelle de l’illustration ci-dessous résultant de cette comparaison est en codons. On voit que jusqu’au codon 32 les deux séquences sont identiques ; ensuite il y a une région de l’allèle muté qui ne se trouve pas dans la séquence de référence. Après, les deux séquences redeviennent identiques. La mutation réside donc dans une insertion de nucléotides dans la séquence de l’allèle de référence.
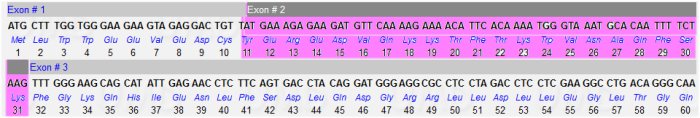
Il reste à identifier les caractéristiques de cette insertion d’une vingtaine de codons. En regardant attentivement l’illustration précédente obtenue avec Anagène, on constate que la séquence ajoutée dans l’allèle muté est exactement identique à celle de l’exon 2 qui la précède. La mutation est donc une duplication de l’exon 2 du gène DMD. On peut s’en rendre compte en considérant la séquence des 3 premiers exons de l’allèle « normal » fourni par la banque.
Il faut comprendre comment la duplication de cet exon 2 est cause de la myopathie du garçon. Pour cela, on peut traduire l’allèle « normal » et l’allèle muté.
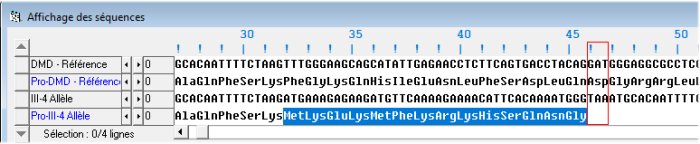
La comparaison des deux protéines montre que la dystrophine mutée a une séquence considérablement réduite par rapport à la dystrophine codée par un allèle normal (45 acides aminés contre 3685). Cette dystrophine mutée est donc non fonctionnelle ce qui entraîne au cours du temps une dégénérescence des fibres musculaires.
Il faut expliquer l’apparition d’un codon stop (TAA) très tôt en position 46 dans la séquence de l’allèle muté. L’exon 1 comprend 31 nucléotides soit 10 codons et un nucléotide. L’exon 2 comprend 62 nucléotides soit 20 codons et deux nucléotides. Le codon 11 est constitué par le dernier nucléotide (T) de l’exon 11 et les deux premiers nucléotides (AT) de l’exon 2. En conséquence la séquence protéique codée par l’ensemble exon1-exon2 comprend 31 acides aminés et est la même dans l’allèle muté et l’allèle normal. Dans l’allèle normal, le codon 32 est constitué par les 3 premiers nucléotides de l’exon 3 (TTT) et code pour l’acide aminé phénylalanine.
Dans l’allèle muté, le codon 32 est constitué par les 3 premiers nucléotides du duplicata de l’exon 2 (ATG) qui code pour la méthionine. Les deux protéines commencent à avoir une séquence différente et 14 codons plus loin apparaît dans l’allèle muté un codon stop.
Bien qu’il n’existe pas d’autre cas connu de myopathie dans la famille, le généticien a proposé de déterminer les génotypes d’autres membres de la famille, en particulier ceux de la mère II5 et du père II4 ainsi que ceux des deux soeurs du garçon myopathe. Cela lui apparait d’autant plus nécessaire que la mère II5 est à nouveau enceinte et que le couple désire connaître le risque d’avoir à nouveau un enfant myopathe. La comparaison des séquences des allèles de ces membres de la famille avec la séquence de référence a permis d’établir le tableau suivant.

La première conclusion à en tirer est que tous les membres diagnostiqués de la famille autres que le garçon III4, en particulier la mère II5, n’ont que des allèles « normaux ». Le génotype de la mère établi à partir des cellules somatiques est :
Xm+/Xm+ et celui du père Xm+/Y .
Le chromosome X du garçon III4 lui a été fourni par sa mère. Pour expliquer qu’elle ait pu transmettre à son fils un allèle cause de sa myopathie, il faut imaginer une mutation du gène DMD (une duplication de l’exon 2 d’un allèle porté par un des chromosomes X de son caryotype) lors de la formation de l’ovule à l’origine, après fécondation, du garçon III4. La notion de néo-mutation (= mutation de novo) est ainsi établie et on peut souligner qu’elle est, bien que rare (10-4) plus fréquente que pour les autres gènes en rapport avec la longueur du gène DMD
Il reste à discuter du risque qu’a le couple II4-II5 d’avoir un second garçon myopathe.
Pour cela, il faut qu’une nouvelle néo-mutation apparaisse lors d’une division intervenant lors de la formation de l’ovule. La mutation étant un phénomène aléatoire, sa probabilité est égale à celle de la fréquence des néo mutations du gène DMD soit 10-4.
La description complète d'un cas de cette mutation dans la banque UMD-DMD est la suivante.
Ces deux myopathies sont phénotypiquement différentes, la myopathie de Becker se traduisant par des symptômes nettement moins sévères que la myopathie de Duchenne. Pourtant elles sont dues à des mutations d’un même gène, le gène DMD codant pour la dystrophine. On peut alors penser que c’est la nature des mutations et plus précisément leurs conséquences au niveau protéique qui sont responsables des différences entre les deux types de myopathies. C’est ce que permettent de tester les documents de cette partie.
On dispose du fichier de séquences DMD-BMD Généalogie familles 3 et 4.edi. Ce fichier comprend 3 séquences : DMD référence ; garçons famille 3 ; garçons famille 4.
Garçons famille 3 est relatif à l’allèle du DMD possédé par les garçons atteints de la myopathie de Duchenne de la famille 3.
Garçons famille 4 désigne l’allèle du gène DMD possédé par les garçons de la famille 4 atteints de la myopathie de Becker. Ce sont donc les séquences de deux allèles mutés à l’origine de deux myopathies différentes.
La comparaison des séquences des allèles mutés avec celle de l’allèle de référence indique que les deux séquences mutées sont plus courtes que celle de l’allèle de référence. Cela indique que les deux mutations sont des délétions.
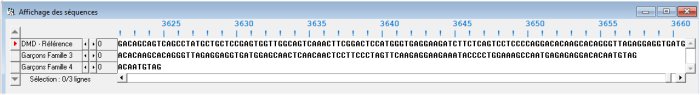
Nature de la mutation de l’allèle de la famille 3
La mutation est une délétion qui débute au codon 765 et se termine au codon 794. Après, les deux séquences sont identiques jusqu’au codon stop ATG terminal.

On peut mettre en évidence la localisation de cette délétion dans la séquence codante complète du gène. Il s'agit de l'exon 19.
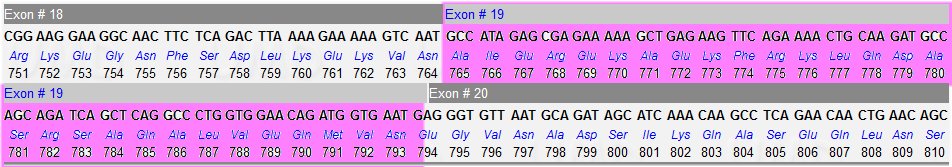
Conséquences de la délétion de 30 codons sur la dystrophine codée par l’allèle muté
Pour cela, il faut traduire les séquences de l’allèle normal et de l’allèle muté puis comparer les séquences des protéines obtenues ; pour cela utiliser la fonction « Comparaison simple » d’Anagène.
La comparaison de l'allèle de référence avec l'allèle des garçons de la famille 3 met en évidence l’absence dans ce dernier d'une région présente dans l’allèle normal.
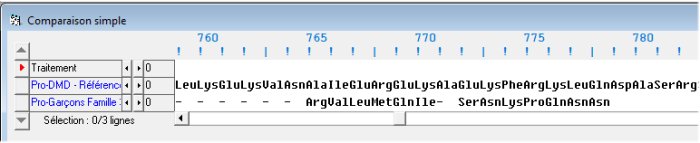
On constate que la protéine codée par l’allèle muté est différente de l’allèle normal à partir de l’acide aminé 765 (ce qui est normal puisque c’est à ce codon que débute la délétion). Mais bien que la séquence de l’allèle muté se prolonge jusqu’au codon stop terminal, le fait le plus frappant est que la protéine mutée est beaucoup plus courte (elle s’arrête au codon 778). En outre, la séquence comprise ente les acides aminés 765 et 778 des protéines « normale » et mutée est différente. La délétion de l’exon 19 provoque un changement dans la séquence de la protéine à partir de l’acide aminé 765 et l’apparition d’un codon stop en position 779.
On peut le vérifier en faisant une comparaison simple des séquences de l’allèle de référence et de l’allèle muté.
On constate l’apparition dans la séquence de l’allèle muté du codon stop TGA en position 779.

L’explication de la différence entre la séquence de l’allèle muté et celle de l’allèle de référence est la suivante : dans l’allèle de référence, la séquence des 18 premiers exons se prolonge par la séquence de l’exon 19. Dans l’allèle muté la séquence des 18 premiers exons se prolonge par celle de l’exon 20 puisque l’exon 19 est délété. Comme les séquences des exons 19 et 20 sont différentes, la succession des codons (et des acides aminés dans la séquence protéique) est différente et un codon stop apparaît dans la séquence de l’allèle muté.
La banque UMD-DMD donne pour l'un des cas signalés le descriptif suivant.
Garçons famille 4
La comparaison avec discontinuités des séquences codantes de l’allèle de référence et de l’allèle des garçons de la famille4 indique que la séquence codante de l’allèle muté est plus courte (3624 codons au lieu de 3685) et que cela est dû à l’absence dans sa séquence d’une région présente dans l’allèle normal. Cette délétion débute au codon 2305 (GTT) et se termine au codon 2366 (AAG). Ensuite, jusqu’au codon terminal, les deux séquences sont identiques. La mutation est donc une délétion de 62 codons.
Début de la délétion
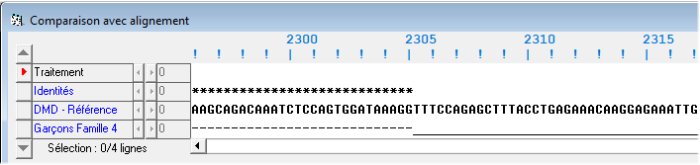
Fin de la délétion (avec imperfection de l'algorithme d'alignement)
La recherche de l’endroit où se trouve située cette délétion dans la structure en exons et introns du gène (séquence codante complète du gène) révèle que c'est l’exon 48 qui est délété.

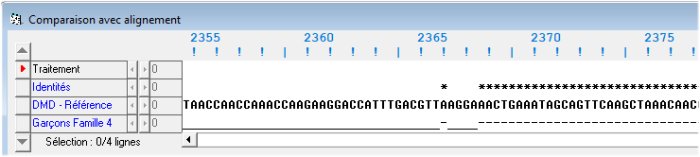
La mutation est une délétion de 62 codons (2305-2366). On peut étudier les conséquences de cette délétion sur la protéine.
Début de la délétion

Fin de la délétion
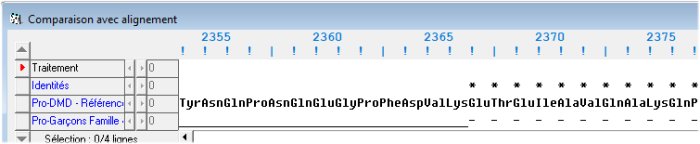
La protéine mutée est plus courte que la dystrophine fonctionnelle. Mais les acides aminés délétés sont uniquement ceux codés par l’exon 48. Au-delà de la délétion, les deux séquences sont identiques. Cela signifie que la délétion de l’exon 48 n’a pas entraîné d’apparition d’un codon stop anticipé dans la séquence de l’allèle muté.
La banque UMD-DMD donne pour l'un des cas signalés le descriptif suivant.
Comparaison des effets des délétions des exons 19 et 48 sur le phénotype de la myopathie.
Les conséquences des délétions des exons 19 et 48 sur la dystrophine sont différentes.
La délétion de l’exon 19 entraîne la synthèse d’une protéine considérablement raccourcie, non fonctionnelle. En particulier il manque toute la région qui normalement se lie aux protéines membranaires. Il en résulte que l’absence de dystrophine fonctionnelle s’accompagne peu à peu d’une dégénérescence des fibres musculaires.
La délétion de l’exon 48 provoque certes un raccourcissement de la dystrophine mais nettement moindre qu’avec la délétion de l’exon 19. En particulier, les régions de la protéine qui se lient à l’actine intracellulaire d'une part et aux protéines membranaires d’autre part sont préservées. La région centrale de la dystrophine, partiellement délétée, n’altère pas complètement la fonction de la protéine.
Cela explique que la délétion de l’exon 19 entraîne les signes cliniques de la myopathie de Duchenne, alors que celle de l’exon 48 s’accompagne des symptômes moins sévères de la myopathie de Becker.
Il reste à expliquer pourquoi les délétions d’exons ont des conséquences différentes sur la protéine. La séquence de l’exon 19 fournie par la banque indique que cet exon comprend 88 nucléotides ce qui n’est pas un multiple de 3. Il comprend 29 codons et un nucléotide (qui formera un codon avec les deux premiers nucléotides de l’exon 20). Il en résulte que la délétion de l’exon provoque un décalage du cadre de lecture de la séquence de l’ARNm au-delà de l’exon 19, à l’origine d’un codon stop anticipé.
L’exon 48 comprend 186 nucléotides, nombre qui est un multiple de 3, et donc 62 codons. Sa délétion n’entraîne pas de décalage du cadre de lecture de l’ARNm en aval. Et les extrémités de la protéine sont préservées.
6. Cas d’une famille où une fille est atteinte de la myopathie de Duchenne
Cette dernière activité nécessite de réinvestir les notions précédemment acquises sur la myopathie ainsi que celles relatives aux accidents génétiques qui peuvent survenir au cours de la méiose. La localisation chromosomique du gène DMD est supposée connue ainsi que la structure de ce gène.
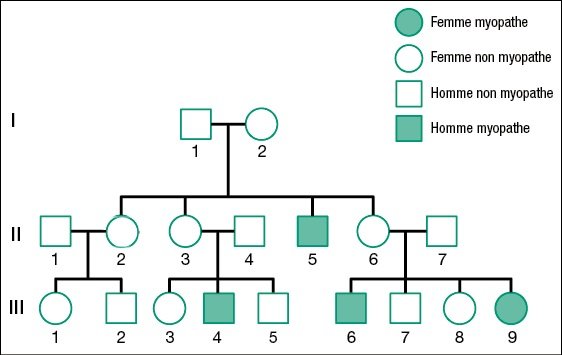
- Le premier point est de dégager les caractéristiques de l’allèle muté qui est le seul allèle possédé par les garçons myopathes III4 et III6. Ayant détecté la nature de la mutation, le généticien a recherché le génotype d’autres membres de la famille, notamment de la fille III9. Le tableau ci-dessous renseigne sur les génotypes des membres de cette famille.
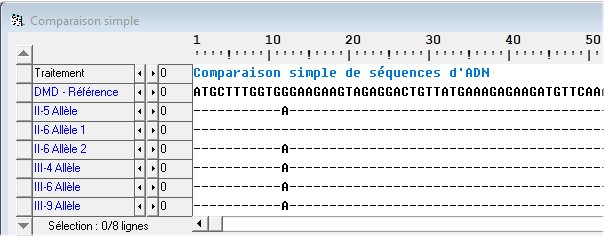
Les données indiquent que les garçons myopathes de cette famille ainsi que la fille III9 possèdent le même allèle muté. Cela témoigne d’une transmission de cet allèle de génération en génération.
Les conséquences de la mutation sur la dystrophine codée par l’allèle muté
On constate une seule différence entre les deux séquences. Le douzième nucléotide à guanine de l’allèle « normal » est remplacé par un nucléotide à adénine (A) dans les allèles mutés. Cette substitution intervient donc précocement. Elle est localisée dans le premier exon et fait que le quatrième codon TGG de l’allèle « normal » est remplacé par le codon TGA de l’allèle muté. Or ce codon TGA est un codon stop de sorte que la dystrophine codée par cet allèle muté ne comprend que 3 acides aminés et est détruite. L’absence de dystrophine fait que la maladie chez les membres de cette famille est une myopathie de Duchenne.
- Le deuxième point est de situer dans le temps l’origine de cet allèle muté. Les données fournies indiquent que la femme II6 est hétérozygote. Elle a donc pu transmettre son allèle muté à son garçon III6 et à sa fille III9. De même la femme II3 qui a un fils myopathe doit être hétérozygote. Les femmes II3 et II6 doivent avoir reçu leur allèle muté de leur mère I2.
Deux solutions sont alors possibles : I2 est elle-même hétérozygote, ou elle est homozygote possédant deux allèles normaux, mais une néomutation est intervenue lors de la formation des gamètes à l’origine, après fécondation, des femmes II3 et II6. Comme la femme I2 a aussi un fils myopathe (II5), la deuxième solution est très peu probable étant donné la faible fréquence des néomutations, même dans le gène DMD.
La femme I2 non séquencée étant hétérozygote, l’allèle muté qu’elle possède lui a été transmis par sa mère. L’origine de l’allèle muté est à rechercher dans les antécédents familiaux non fournis par l’arbre et suppose une néomutation chez une femme.
- Le troisième point à expliquer est la myopathie de la fille III9. Son père n’étant pas myopathe, elle doit avoir reçu un chromosome X de son père possédant l’allèle « normal ». Sa mère étant hétérozygote elle a pu recevoir un chromosome X porteur de l’allèle muté de sa mère. Elle peut donc avoir le génotype Xm+Xm ; étant donné la dominance de l’allèle « normal », elle ne devrait pas être myopathe. Cela est contraire à ce qu’on observe. Une première explication consisterait à dire que chez cette fille, l’allèle muté n’est pas récessif mais dominant sans qu’on en connaisse les raisons. Le document fourni conduit à une autre explication. Le caryotype de cette fille montre qu’elle ne possède qu’un seul chromosome sexuel, le chromosome X.
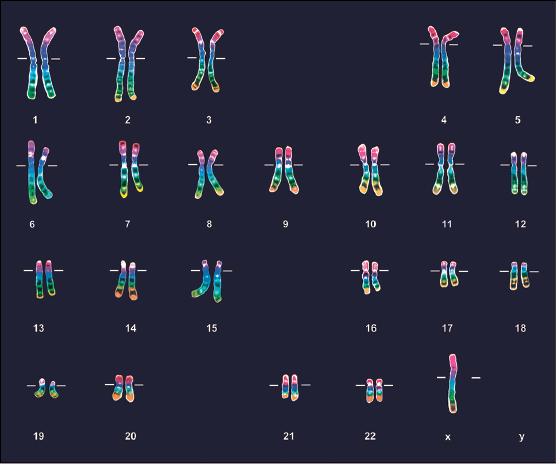
C’est ce chromosome qui doit posséder un allèle muté. Il ne peut provenir que de sa mère. En conséquence le spermatozoïde de son père (II7) ne lui a pas fourni de chromosome X. Lors de la méiose à l’origine de ce spermatozoïde, il y a eu une anomalie. Par exemple, à la première division de la méiose, les deux chromosomes sexuels ont été vers le même pôle de la cellule de sorte qu’une des deux cellules résultant de cette division ne possédait pas de chromosome sexuel.
La myopathie de cette fille III9 résulte donc du fait qu’elle a reçu de sa mère hétérozygote un chromosome X porteur de l’allèle muté et aucun chromosome sexuel de son père.