
L'homme à utérus
Rédigé par Françoise Jauzein, ACCES 203
Relue par Nathalie Josso, INSERM U293
Révisé en 2018
Nommé aussi syndrome de persistance des canaux de Müller (PMDS), pour "Persistent Müllerian Duct Syndrome", ce phénotype correspond à la présence d'un utérus et de trompes chez des individus normalement virilisés. Peu de cas sont connus, environ 150 ont été décrits dans la littérature.
A la naissance, l'enfant est sans ambiguité déclaré de sexe masculin, il possède un caryotype 46,XY. Ce n'est souvent qu'à l'occasion d'une intervention chrirurgicale programmée à cause d'un cryptorchidisme unilatéral ou bilatéral que ce phénotype particulier est découvert.
Les anomalies anatomiques
Il existe deux formes anatomiques de PMDS.
Dans la grande majorité des cas (80%), le cryptorchidisme est unilatéral et associé à une hernie inguinale contralatérale. Un des testicules est descendu dans le scrotum, et l'utérus ainsi que la trompe de Fallope située du même côté sont entrés dans le canal inguinal (ce que l'on appelle une hernie utérine inguinale) ou peuvent y glisser facilement sous l'effet d'une légère traction. L'utérus et la trompe de Fallope situés de l'autre côté sont alors déplacés dans l'espace vacant. Le testicule opposé est souvent en position de hernie inguinale, ce que l'on nomme "ectopie testiculaire transverse" (les deux testicules sont dans la même bourse).
Plus rarement (20%), le cryptorchidisme est bilatéral, l'utérus est fixé dans le pelvis et les deux testicules sont inclus dans le ligament rond.
|
|
| Comparaison des anatomies internes d'une femme normale (à gauche) et d'un enfant mâle atteint de PMDS (à droite). Dessin de droite d'après Loeff et al. A noter : la liaison des testicules avec les trompes de Fallope ainsi que la proximité des canaux déférents avec l'utérus. |
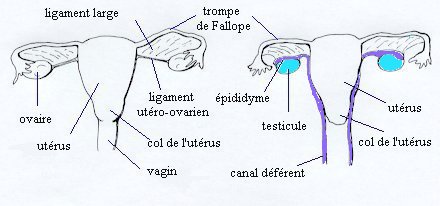
Puisque, dans ce syndrome les testicules sont étroitement attachés aux trompes, leur descente dans les bourses est liée à la mobilité de ces dérivés Müllériens.
Une dégénérescence progressive du tissu testiculaire a été parfois observée. Elle est due à une torsion du testicule en relation avec sa grande mobilité dans ce syndrome. En effet, il n'est ni attaché aux processus vaginaux, ni à la base du scrotum par le Gubernaculum.
Si le cryptorchidisme n'est pas trop ancien, les testicules peuvent présenter des cellules germinales mais ils ne sont que rarement correctement connectés avec leurs canaux excréteurs. Ils ne seront donc pas fonctionnels.
L'origine hormonale du syndrome
La différenciation sexuelle normale est contrôlée par deux hormones testiculaires, l'hormone anti-Müllérienne et la testostérone. En absence de "signal" AMH, les canaux de Müller évoluent en utérus, trompes de Fallope et partie supérieure du vagin, alors que le manque de "signal" testostérone entraîne l'évolution des organes génitaux externes et du sinus urogénital vers un phénotype femelle.
Le PMDS correspondrait donc à un défaut du signal AMH.
Deux hypothèses se présentent alors : soit cette hormone est absente, soit il n'existe pas de récepteurs normaux sur ses cellules cibles.
Les individus atteints du syndrome de persistance des canaux Müllériens ont des taux de testostérone normaux, et répondent normalement à des stimulations par l'hCG (hormone de type LH mais à durée de vie plus longue) par une augmentation de la sécrétion de testostérone. Cependant, certains d'entre eux, présentant une dégénérescence du tissu testiculaire vraisemblablement due à une torsion testiculaire précoce, sont insensibles à l'hCG.
Chez les garçons normaux, le taux d'AMH est détectable de la naissance à la puberté puis baisse brusquement à cette époque en raison de l'inhibition exercée sur l'expression du gène de l'AMH par la testostérone, dont le taux augmente fortement.
Chez les individus atteints de PMDS, l'analyse des taux sériques d'AMH fournit des résultats hétérogènes. Dans environ la moitié des cas, les taux d'AMH sont normaux pour l'âge ou à la limite supérieure. Dans l'autre moitié, ces taux sont très bas ou même indétectables.
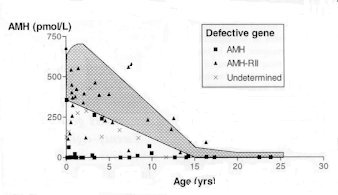 |
| Taux d'AMH sérique chez l'homme en fonction de l'âge. Avant 15 ans, un PMDS chez un individu présentant un taux normal d'AMH (zone grisée) correspond à une déficience du recepteur de type II (sujet AMH-positif) alors qu'un taux faible indique une déficience dans l'AMH elle-même (sujet AMH-négatif). |
La première moitié des cas correspond à des individus qui présentent une mutation des récepteurs de type II à l'AMH, ils sont appelés sujets "AMH-positifs". L'autre moitié, correspond à des sujets "AMH-négatifs". Ils présentent le plus souvent une mutation du gène de l'AMH lui-même. Cependant, ce faible taux ne peut guère être interprété après la maturité sexuelle (où le taux d'AMH est physiologiquement bas) ou chez des individus présentant une dégénérescence du tissu testiculaire.
Les mutations du gène de l'AMH
Le gène de l'AMH a été cloné en 1986 par R. Cate. C'est un petit gène de 2.8 kb, situé sur le chromosome 19 (19p13,3), organisé en cinq exons (l'extrémité 3' du cinquième exon, qui code pour la partie C-terminale de la protéine, montre une nette homologie avec la super-famille des facteurs de croissance et de transformation de type TGF-bêta, conservée dans de nombreuses espèces).
Tous les membres de cette famille sont des glycoprotéines dimériques, liées par des ponts dissulfures, qui sont clivés au niveau d'un site situé de 110 à 125 acides aminés de l'extrémité C-terminale de la protéine, pour libérer le facteur de croissance mature. Le segment C-terminal du TGF-bêta doit être séparé du segment N-terminal pour être actif. De la même façon, l'activité biologique de l'AMH est due à sa partie C-terminale (mais une association non covalente avec sa partie N-terminale rehausse son activité au lieu de l'inhiber, vraisemblablement en assurant à la protéine une conformation qui facilite sa sécrétion par le reticulum endoplasmique).
La première mutation du gène de l'AMH a été mise en évidence dans une famille en 1991, par un test biologique (en absence de test ELISA à cette époque) consistant en la mise en culture de biopsies testiculaires des trois garçons avec des tractus de rat indifférenciés. L'absence de régression des canaux de Müller des rats prouvant l'absence d'AMH dans les sécrétions testiculaires humaines. Il s'agissait d'une mutation STOP située dans le cinquième exon et entraînant la perte des derniers 178 acides aminés, incluant la portion C-terminale biologiquement active de l'AMH.
|
|
| Le gène de l'AMH et ses mutations.
N. Josso, J-Y. Picard, S. Imbeaud, N. di Clemente et R. Rey : Clinical aspects and molecular genetics of the persistent Müllerian duct syndrome, Clinical Endocrinology, 1997, 47, p137-144 Les 5 exons sont en jaune et les introns en blanc. Les mutations STOP et les délétions (numéro de nucléotides manquant) sont indiquées au dessus de la séquence, le polymorphisme est localisé en pointillé, les mutations non-sens sont indiquées en dessous. La séquence est celle correspondant à l'ARNm (le brin codant), les acides aminés (une lettre) modifiés sont localisés. |
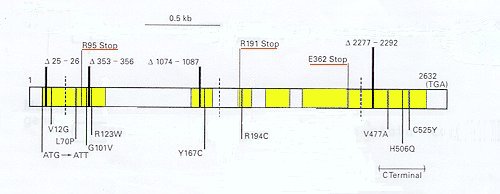
Les mutations du gène de l'AMH sont très variées. Les plus courantes sont trois mutations STOP ( interrompant la traduction de l'ARNm), quatre petites délétions et 11 mutations faux-sens (l'une d'entre elles transformant le codon initiateur ATG en ATT). Enfin, le polymorphisme de l'AMH est important, environ 35% des allèles diffèrent de l'allèle de référence.
Mutations du gène du recepteur de type II de l'AMH
Le récepteur primaire, nommé type II, fixe l'AMH de façon indépendante mais nécessite la présence du récepteur de type I pour entraîner la réponse biologique de la cellule cible. Aujourd'hui, seul le récepteur de type II a été cloné, d'abord chez le rat puis le lapin. Le gène humain du récepteur de type II mesure 8.2 kb, arrangés en 11 exons, et ne subit pas d'épissage alternatif (comme c'est le cas chez le lapin où un épissage alternatif amène à un transcrit court, dépourvu d'exon 2, et donnant un récepteur incapable de lier l'AMH). Les exons 1 à 3 codent pour une partie extracellulaire du récepteur, l'exon 4, pour sa portion transmembranaire. La protéine comprend 573 acides aminés et son homologie avec d'autres récepteurs de type II de facteurs de croissance de la famille du TGF-bêta n'excède pas 33%. Ce gène est situé sur le chromosome 12 (12q13).
La première mutation rencontrée dans ce gène est une substitution de G en A au niveau de l'intron 2. Elle entraîne un épissage alternatif au niveau de ce site, certains transcrits ne présentent pas l'exon 2 alors que d'autres sont légèrement plus longs que la normale car l'epissage se fait en un site positionné 12 paires de bases en aval du second intron. Cela implique le changement d'un acide aminé (Gly78 devient Asp) et l'adjonction de quatre acides aminés supplémentaires. Le transcrit court donne un récepteur inactif, comme chez le lapin, et le transcrit long donne une protéine qui reste dans le reticulum endoplasmique.
Les mutations de ce gène sont très diverses, on a trouvé trois mutations STOP, une micro-délétion et 7 mutations faux-sens. Cependant, alors que les mutations du gène de l'AMH sont "propres" à chaque famille, dans le cas du gène de son récepteur II, un grand nombre de patients présentent la même mutation : une délétion de 27 bases située dans le dixième exon, sur l'un ou les deux allèles. Le cadre de lecture du gène reste intact mais 9 acides aminés manquent dans la région intracellulaire de la protéine récepteur. Cette délétion représente au total 25% des cas de PMDS, elle est facile à détecter par la technique de PCR.
|
|
| Mises au point (1999) sur les mutations du gène de l'AMH et de celui du récepteur de type II de cette hormone. Les exons sont grisés. La séquence est celle correspondant à l'ARNm (le brin codant). Les acides aminés (une lettre) modifiés sont localisés. Par exemple, pour la "dernière" mutation faux-sens du gène de l'AMH : le 525°acide aminé, une Cystéine (C), est remplacée par une Tyrosine (Y). Les mutations faux-sens sont notées au dessus de la molécule. Les mutations non-sens, les insertions ou les délétions sont notées en dessous. Les astérisques représentent des mutations de sites d'épissage. Les mutations récurrentes sont entourées. |

Mode de transmission du PMDS
Une transmission autosomale du phénotype avait été supposée, elle a été confirmée par la localisation du gène de l'AMH sur le chromosome 19 et celui du récepteur de type II sur le chromosome 12.
Les études au sein des familles montrent le plus souvent une récessivité de la transmission, tous les patients déficients en AMH présentent deux allèles mutés ainsi que la majorité des patients présentant un PMDS par défaut de récepteurs de type II.
Les mutations du gène de l'hormone anti-Müllérienne se rencontrent essentiellement dans les populations arabes des régions méditerranéennes caractérisées par un fort taux de consanguinité (81% des patients sont homozygotes). Par contre, les patients présentant des mutations du gène du récepteur II de l'AMH sont le plus souvent originaires du nord de l'Europe, et seulement 45% sont homozygotes.
Sources
- C. Belville, N. Josso and J-Y. Picard : Persistence of Müllerian Derivatives in Males, in American Journal of medical Genetics (Semin. Med. Genet.) 89:218-223, (1999)
- N. Josso, J-Y Picard, S.Imbeaud, N. Di Clemente and R.Rey : Clinical aspects and molecular genetics of the persistent Müllerian duct syndrome, in Clinical Endocrinology 47:137-144 (1997).