
AVC : aspects cellulaires et moléculaires
I- La notion de pénombre ischémique
a- Débit sanguin cérébral normal et pathologique
Le débit sanguin cérébral est en moyenne de 50mL/min/100g de tissu cérébral chez l'adulte. Ce débit varie en fonction de l'âge, de l'état de santé et de l'activité cérébrale. L'infarctus cérébral est la conséquence de la diminution puis de l'arrêt de la perfusion et du dépassement des capacités des systèmes de suppléance c'est à dire des systèmes capables de contrer cet arrêt de la perfusion comme par exemple une autorégulation du débit sanguin cérébral. Ces systèmes de protection sont mis en jeu dès que le débit passe sous la valeur de 50mL/min/100g de tissu cérébral. Pour 20mL/min/100g de tissu cérébral, le métabolisme tissuaire est altéré et des symptômes neurologiques peuvent apparaître. En deça de 15mL/min/100g de tissu cérébral, on parle de pénombre ischémique, ce qui se traduit par une activité électrique nulle à l'électroencéphalogramme, réversible à condition que le flux sanguin soit rétabli. Par contre, si cet état se prolonge, on passe en quelques minutes au stade de nécrose tissulaire (elle survient concrètement pour un débit inférieur à 10mL/min/100g de tissu cérébral pendant plus de 3 min). Figure 1.
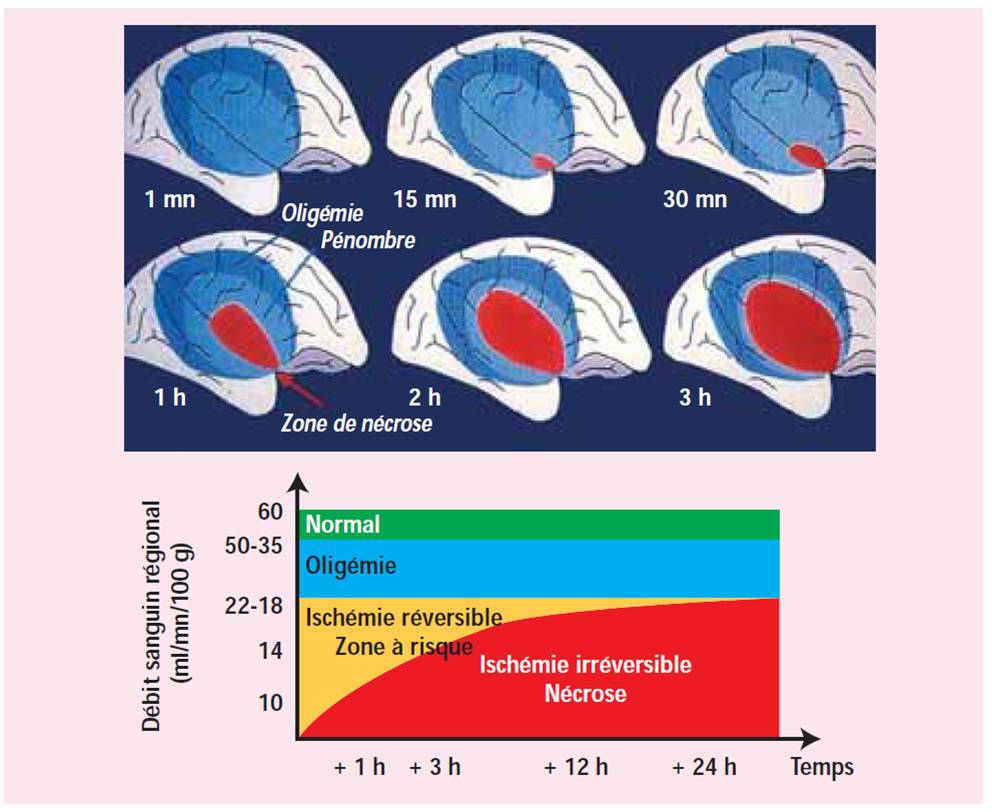 |
| Figure 1. Représentation schématique de la pénombre (N. Nighoghossian, Correspondance en neurologie vasculaire, Vol. 5 n°2, oct-dec 2005) |
b. Méthodes d'évaluation
On dispose de plusieurs approches pour évaluer ces variations de perfusion :
- Tomographie à émission de positons (TEP) : c'est l'outil le plus pertinent car il permet d'accéder à des variables comme le débit sanguin, la consommation d'oxygène, le taux d'extraction de l'oxygène au sein de la zone d'infarctus et en périphérie.
- Imagerie par résonnance magnétique (IRM): par IRM de diffusion et de perfusion, il est possible de réaliser une évaluation précoce des dommages tissulaires. C'est le concept de mismatch (qui est encore discuté et affiné), fondé sur la soustraction des volumes de perfusion et de diffusion qui permet d'identifier en urgence la zone à risque (figure 2) : la zone de nécrose est assimilée au volume lésionnel objectivé par les séquences de diffusion.
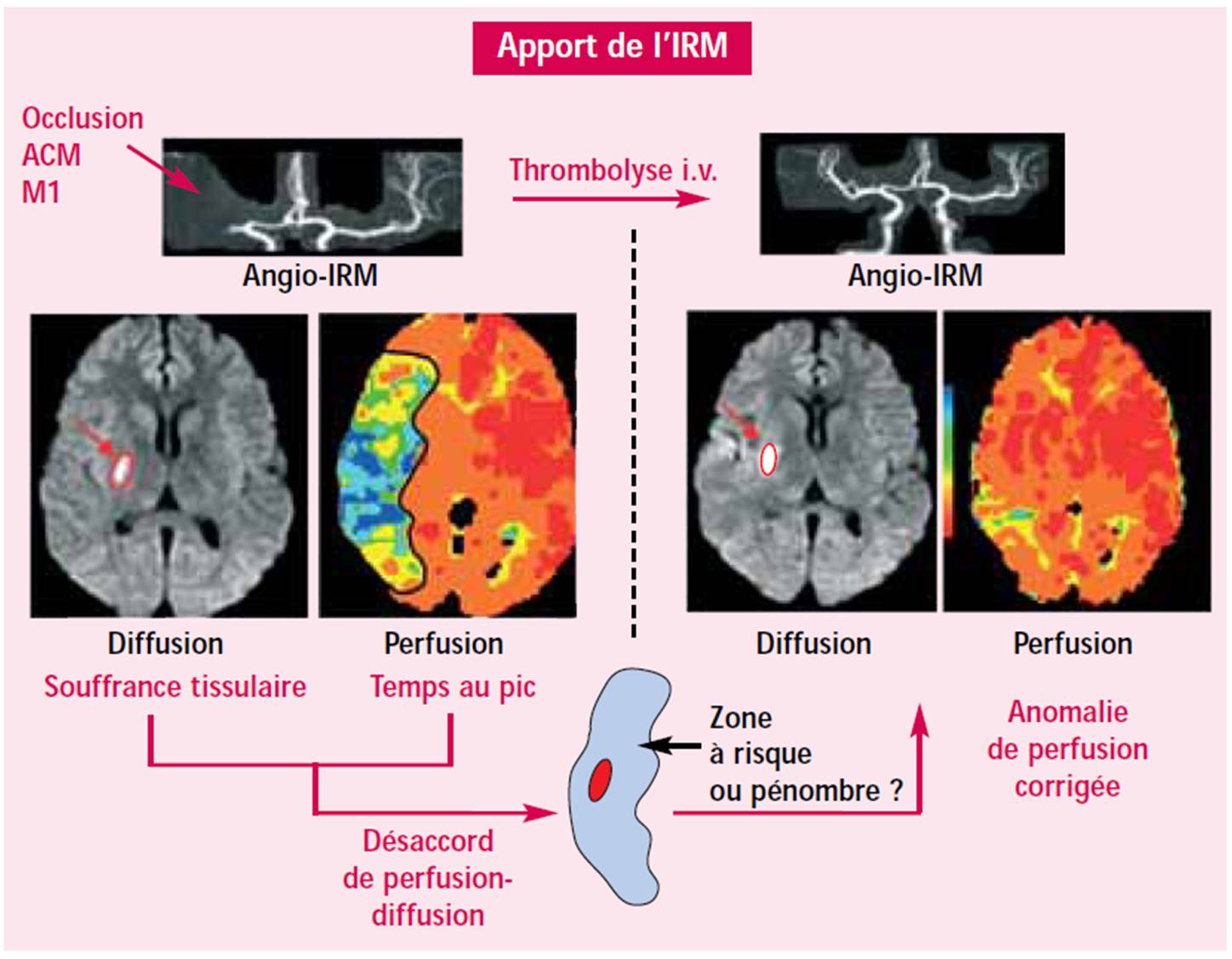 |
| Figure 2: Correction du mismatch en IRM après recanalisation par thrombolyse (N. Nighoghossian, Correspondance en neurologie vasculaire, Vol. 5 n°2, oct-dec 2005) |
- Scanner : des études de scanner couplé au Xénon permettent une évaluation des zones de nécrose et de pénombre.
II- L'excitotoxicité, une conséquence de l'ischémie cérébrale
a- Les multiples conséquences de l'ischémie
Les lésions tissulaires consécutives à une ischémie ne s'expliquent pas seulement par la privation en métabolites énergétiques et en oxygène. En effet les lésions sont la résultante de mécanismes complexes variant dans le temps et l'espace.
Le coeur de l'ischémie est le siège d’une nécrose, souvent rapide et secondaire à des processus de mort cellulaire d’origine cytoplasmique où la libération massive de calcium joue un rôle important. En revanche, la zone de pénombre, celle-là même où persiste une certaine perfusion, est le siège de l’apoptose, un processus de mort cellulaire. La maturation des lésions ischémiques est par ailleurs la résultante de la mise en oeuvre d’une succession de processus délétères débutant immédiatement au décours de l’ischémie, tels que l’excitotoxicité ou le stress oxydant, et se prolongeant parfois plusieurs jours, comme l’inflammation post-ischémique. Figure 3.
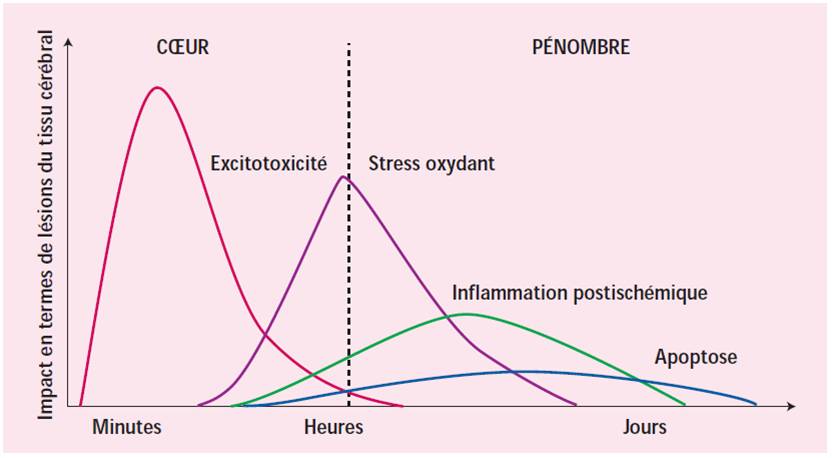 |
| Figure 3: Evolution spatio-temporelle des mécanismes impliqués dans l'ischémie cérébrale (Correspondances en neurologie vasculaire Vol. VI, jan 2006, Deplanque) |
Au cours du temps, il existe donc un recrutement progressif de la zone de pénombre (c'est à dire d'altérations neurologiques potentiellement réversibles) par le foyer ischémique (c'est à dire de mort neuronale). Cette évolution est contrôlée par une cascade d'événements qui évoluent dans le temps et l'espace et qui sont l'excitotoxicité, l'apoptose et l'inflammation.
b- Calcium et excitotoxicité
La conséquence la plus rapide et la plus importante de l'ischémie est l'augmentation de la concentration intracellulaire de calcium pour deux raisons principales:
- la déplétion énergétique (car l'ATP est indispensable pour soutenir l'activité des protéines membranaires maintenant la concentration intra-cellulaire en calcium très faible (de l'ordre de 0,1 micromol/ L)),
- la libération massive d'acides aminés excitateurs comme le glutamate. Cette libération massive s'effectue de manière incontrôlée suite à la dépolarisation neuronale consécutive au stress. La stimulation excessive des récepteurs au glutamate (surtout les NMDA) accroît et entretient l'augmentation intracellulaire de calcium dans la cellule.
Le calcium met alors en jeu plusieurs cascades enzymatiques aboutissant entre autres à l'inhibition de la synthèse protéique et la production de radicaux libres: cela aboutit à la destruction de la cellule et donc à la nécrose.
C- NO, stress oxydant et ischémie cérébrale
Le NO apparaît aussi comme un acteur impliqué après une ischémie et il présente un double visage :
- délétère s'il est produit en raison de l'élévation du calcium intra-cellulaire par la NOsynthase I dans les 10 premières minutes de l'ischémie, puis par la NOsynthase II des cellules astrocytaires et gliales dans les 12 h suivantes. Dans ce cas, c'est l'aspect radicalaire du NO qui conduit à des réactions radicalaires en chaîne: il se produit un stress oxydant.
- bénéfique s'il est issu de l'activité de la NOsynthase III endothéliale dès la première heure de l'ischémie. Il diminue alors l'agrégation plaquettaire, contrôle le tonus vasculaire et améliore le débit sanguin cérébral.
Le stress oxydant provoque des effets délétères majeurs dans le système nerveux car les neurones ont une mauvaise défense anti-oxydants. Or, l'ischémie et le glutamante conduisent la mitochondrie et surtout la NADPH oxydase à génèrer des espèces réactives oxygénées (ROS) comme l'anion superoxyde.
D- Inflammation post-ischémique
Le cocktail calcium, NO, stress oxydant conduit à l'activation de facteurs de transcription (comme NFkB) qui accroissent les effets délétères:
- augmentation de la synthèse de NOsynthase donc de NO
- expression de la cyclo-oxygénase 2 ce qui provoque l'apparition de prostanoïdes toxiques et oxydatifs
- expression de nombreuses cytokines pro-inflammatoires recrutant, activant et regroupant des cellules gliales et des macrophages, ce qui majore les lésions ischémiques.
La production de facteurs pro-inflammatoires présente malgré tout un effet neuroprotecteur: par exemple le TGF béta limite l'extension des lésions en se fixant sur des récepteurs des neurones et des astrocytes. Ces dernières cellules libérent en réponse au TGF une protéine inhibitrice de l'activateur de plasminogène (PAI-1): alors que l'activateur tissulaire du plasminogène (t-PA) stimule la mort neuronale, le PAI-1 joue le rôle exactement inverse. Cette protéine se fixe aux récepteurs NMDA qu'elle clive partiellement et limite ainsi l'entrée de calcium dans les cellules.
E- Apoptose
L'apoptose va compléter plus tardivement ce tableau négatif et ce processus cellulaire se retrouve surtout dans la zone de pénombre.
Les caspases sont des protéases intra-cellulaires à cystéine au site actif clivant leur substrat après un aspartate: ces enzymes participent à l'induction, l'amplification et l'exécution de l'apoptose. Il a été montré que ces enzymes sont actives dans les neurones et les oligodendrocytes dans le coeur et la pénombre après ischémie focale. L'utilisation expérimentale d'inhibiteurs des capases comme des tétrapeptides de synthèse mimant le substrat réduit la perte neuronale mais ne peut encore pas annuler les déficits mnésiques induits par la lésion (Gillardon et al, Brain Res, 1996, 40, 254-260). Ainsi, les lésions aiguës du système nerveux central apparaissent ainsi en bonne position pour bénéficier d’une ouverture thérapeutique anti-apoptotique à une échéance qu’il est encore difficile de préciser, mais qui n’appartient plus au domaine de la fiction.
A propos de l'apoptose, il est très important de présenter les rôles des microparticules apoptotiques. En effet, des plaques carotidiennes d'athérome sont présentes chez 40 à 50% des patients ayant un infarctus cérébral et en sont la cause directe chez 10 à 30% des cas environ: la rupture de la plaque peut conduire à une obstruction complète de certaines artères à cause du développement d'un énorme thrombus. Or, dans ces plaques, se trouvent des cellules apoptotiques contribuant aux phénomènes inflammatoires conduisant à la rupture de la plaque: les microparticules apoptotiques relarguées pourraient servir de marqueurs sanguins annonciateurs du la rupture.
→ L'apoptose et les microparticules apoptotiques
L'apoptose (ou mort cellulaire programmée; MCP) est définie par des critères précis à l'échelle cellulaire et moléculaire (condensation du cytoplasme, condensation de la chromatine, fragmentation de l'ADN...). La formation de corps apoptotiques ou microparticules apoptotiques (MPA) est un des événements les plus remarquables: il s'agit de "fragments" (entités entourées de membrane plasmique et contenant du cytoplasme modifié) de la cellule apoptotique qui seront éliminés par des macrophages.
Plusieurs données expérimentales indiquent un rôle pro-inflammatoire de l'apoptose dans l'athérosclérose: ainsi, les MPA sont capables d'induire une activité inflammatoire des cellules endothéliales qui se mettent à produire des cytokines (Il 6) et des chimiokines (MCP-1). L'induction et la pérennisation de la réponse immuno-inflammatoire dans la paroi artérielle à l'origine du développement des plaques d'athérosclérose sont donc liées à la production de MPA.
→ L'apoptose, un processus à potentiel coagulant
Des expériences menées in vitro suggèrent que les cellules apoptotiques augmentent aussi la thrombogénèse: ces cellules expriment dans leur feuillet externe plus de phosphatidyl-sérine (PS). Ceci conduit à une augmentation de l'activité du facteur tissulaire (FT): PS et FT favorisent alors la formation des complexes catalytiques de la coagulation donc cela est en faveur d'une propension accrue à la formation de caillots.
D'autre part, la plaque d'athérome contient des MPA, dont la membrane est riche en PS: les plaques d'athérome ont donc une activité pro-coagulante. Lors de la rupture des plaques, il y a libération de nombreuses MPA qui conduisent à la formation de thrombus.
Pour résumer, les MPA provoquent l'apparition des plaques et leur développement dans un contexte inflammatoire. Ces MPA sont ensuite érodées des plaques et présentent un potentiel élevé de thrombogénicité d'où la dissémination et la persistance d'un état procoagulant.
Chez de nombreux patients atteints par des infarctus du myocarde ou des AVC, il a été observé des taux significativement plus élevés de ces MPA circulantes. Ces MPA constituent donc un marqueur fiable de la présence d'une complication thrombogène d'une plaque d'athérome quelque soit sa localisation et pourrait constituer un marqueur pronostic de récidives ischémiques.
Mallat et al, Circulation 1999; 99: 348-53
Mallat et al, Circulation 2000; 101: 841-3
La reperfusion post-ischémique
En considérant l'ischémie vasculaire sous l'angle purement vasculaire, la reperfusion apparaît comme un élément important dans la limitation des lésions neurologiques. Ceci a conduit à l'utilisation de rt-PA (activateur tissulaire du plasminogène recombinant, assurant la transformation de plasminogène en plasmine qui induit la fibrinolyse) dès 1996 aux USA et dès 2002 en Europe.
Les effets bénéfiques de la reperfusion paraîssent évidents pour limiter les lésions neurologiques, mais les effets ne sont pas toujours bénéfiques:
- Le passage brutal d'une hypoxie à une reperfusion majore les processus oxydatifs (donc le stress oxydant) et l'afflux de leucocytes (donc l'inflammation) ce qui accroît les lésions!
- La reperfusion peut provoquer une altération de la réactivité vasculaire (perte du tonus vasculaire) d'où une limitation des possibilités d'auto-régulation du débit sanguin local.
L'altération oxydative du fonctionnement d'un canal potassique Kir 2.1 des cellules musculaires lisses vasculaires (CMLV) réduit la capacité d'auto-règulation du débit sanguin: ces canaux maintiennent le potentiel de repos des CMLV et participent surtout à la vasodilatation. Ces canaux sont activés par une augmentation légère de la concentration extra-cellulaire en K+ ce qui provoque une sortie de K+, donc une hyperpolarisation qui inactive des canaux calciques ce qui conduit à une vasodilatation. Or la concentration en K+ extra-cellulaire est proportionnelle à l'activité des neurones, donc ces canaux participent directement à un contrôle local de la perfusion de manière proportionnelle à l'activité des neurones.
-
Une possible toxicité du rt-PA pour deux raisons : une majoration de l'excitotoxicité par sensibilisation des récepteurs NMDA ainsi qu'une induction d'hémorragies cérébrales.
Conclusion
Le caractère évolutif, la diversité et la complexité des mécanismes mis en jeu au cours de l’ischémie cérébrale permettent de comprendre, dans une certaine mesure, les échecs ou les succès relatifs de certains traitements.
Les interactions entre les systèmes vasculaire et neuroglial apparaissent aujourd’hui comme déterminantes dans le développement des lésions cérébrales consécutives à l’ischémie. À l’avenir, les différentes approches thérapeutiques devront tenir compte de l’ensemble de ces paramètres.