Glossaire
Histoire
Téléchargements
L'enzyme PAH et le cofacteur Bh4
La protéine codée par le gène PAH
La phénylalanine hydroxylase se présente sous forme d'un tétramère. Nous présentons ci-dessous la forme dimérique dans laquelle les 2 monomères sont associés par l'intermédiaire des hélices a du bout C-terminal.
Le repérage du site catalytique est possible en étudiant le complexe enzyme-substrat ou le complexe enzyme-inhibiteur. La structure spatiale du cofacteur (tétrahydrobioptérine) est très proche de celle des inhibiteurs (adrénaline, nor-adréanline, Dopa et L dopa) : ceci permet de comprendre pourquoi l'inhibiteur est capable de prendre la place du cofacteur au niveau du site catalytique de la phénylalanine hydroxylase. Le résultat de cette recherche qui permet le repérage du site catalytique est reproduit dans l'animation ci-dessous : (L'identification du site catalytique peut être effectuée grâce au logiciel Rasmol en utilisant la fonction restrict within (5.0,HBI). Cette fonction permet d'afficher tous les atomes situés dans un rayon de 5.0 nm autour du cofacteur HBI (tétrahydrobioptérine). La même démarche peut être mise en oeuvre à partir du complexe enzyme-inhibiteur).Le site catalytique met en jeu un atome de fer, trois molécules d'eau et trois résidus : Glu330, His290 et His285. Le repèrage du site catalytique peut être réalisé sur les modèles montrant l'enzyme associée :
- soit au cofacteur
- soit à un inhibiteur compétitif : complexe enzyme_adrénaline, complexe enzyme_nor adrénaline, complexe enzyme_dopamine ou complexe enzyme_Ldopamine.
- Le monomère de la phénylalanine hydroxylase.
Bh4 est synthétisée de novo à partir du guanosine triphosphate (GTP) au cours de 4 réactions enzymatiques catalysées par 3 enzymes différentes : GTPCH, PTPS et SR. Mais cette synthèse est insuffisante pour assurer une concentration suffisante en Bh4. Un aspect fondamental du métabolisme de Bh4 est sa régénération à tavers 2 étapes ctalysées par les enzymes PCD et DHPR.
Des mutations de ces gènes, en particulier celui qui code pour l'enzyme DHPR, peuvent entraîner un déficit en Bh4 à l'origine d'une hyperphénylalaninémie. De plus, Bh4 est aussi un cofacteur des enzymes tyrosine hydroxylase et tryptophane hydroxylase qui catalysent des réactions d'hydroxylation à l'origine des neuromédiateurs dopamine (et au delà, de la nor-adrénaline) et sérotonine.
Le déficit en Bh4 se traduit ainsi, en plus d'une hyperphénylalaninémie, par un déficit de la neurotransmission monoaminergique accentuant les signes neurologiques de la maladie.
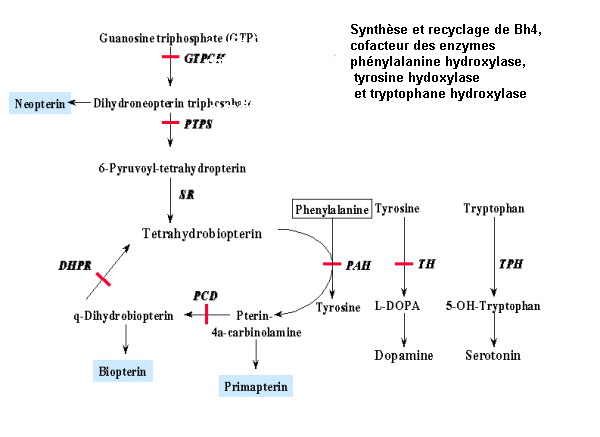
D'après le site de Bh4. Diffusion autorisée.