Glossaire
Histoire
Téléchargements
Rédigée par Jacques Barrère,
Lycée P.L. Courier, Tours
Relue par Henri Wajcman, U468 de l'INSERM
Qu'est-ce que la drépanocytose ?
La drépanocytose est une maladie autosomale récessive due à une mutation unique, ponctuelle, du gène b globine situé sur le chromosome 11 (11p 11-5). La mutation du 6 ème codon (GAG6GTG ) entraîne le remplacement de l'acide glutamique 6 par de la valine (Glu6Val).
En fait, il existe pour la maladie drépanocytaire, différents génotypes : la drépanocytose résulte de la combinaison de 2 allèles du gène de la bêta globine dont au moins 1 porte la mutation Glu6Val. Trois génotypes prédominent : HbS//HbS (70%), HbS//HbC (25%), HbS//Hb-Thalassémie (5%). Les sujets hétérozygotes HbA//HbS sont des porteurs sains. Les hématies sont normalement discoïdes ; celles qui contiennent l'hémoglobine S s'incurvent, se déforment en forme de faucille ébréchée, lorsqu'elles sont désoxygénées. Dans un premier temps cette déformation entraîne le blocage des capillaires, ce qui entraîne des ischémies locales, pouvant être très grave, avec des crises vaso-occlusives particulièrement douloureuses dans les muscles et des risques de complications organiques graves (squelette, rate, tube digestif, cerveau).Voir les séquences nucléiques Voir les séquences protéiques
Autrefois 80% des homozygotes (HBS//HBS) mouraient avant l'âge de la reproduction. Aujourd'hui, grâce au dépistage précoce, à la prévention des infections (vaccination, antibiothérapie systématique), à la prévention de la déshydratation et de tout autre cause pouvant la provoquer, la maladie reste sérieuse et invalidante mais l'espérance de vie s'est considérablement normalisée.
Pourquoi le remplacement Glu 6 Val au niveau de la globine b de l'hémoglobine désoxygénée provoque-t-elle une déformation des hématies ?
La désoxyhémoglobine HbS présente aussi bien in vivo qu'in vitro une propriété nouvelle : la polymérisation. Cette dernière ne se produit qu'en solution concentrée comme c'est naturellement le cas dans l'hématie. Le retour de l'HbS à l'état oxygéné (oxyhémoglobine S) provoque la dissociation des polymères.
On
explique la polymérisation de la désoxyhémoglobine
S (attention image 3D en visualisation dynamique sous Chime : taille
du fichier 110 Ko) par le fait que la valine n°6 est un résidu
hydrophobe qui remplace un acide aminé hydrophile, l'acide glutamique.
Les globines étant entourées par un film d'eau, la présence
d'un site hydrophobe crée un point de "collage" entre 2 molécules
d'hémoglobines voisines. Celui-ci s'établit entre la leucine
88 et la phénylalanine 85 d'une chaîne alpha d'une molécule
d'hémoglobine et la valine 6 de la chaîne b
de l'hémoglobine voisine, d'où création d'une structure
cristalline en fibres.
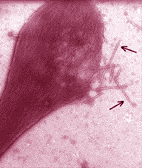 |
Vu au microscope électronique, le globule rouge drépanocytaire désoxygéné apparaît rempli d'un gel formé par des de cristaux allongés longs de 1 à 15 µm. Ces cristaux sont constitués par des polymères d'hémoglobine. Le globule rouge, déformé par ces structures fibreuses tubulaires prend une forme caractéristique en faucille ou feuille de houx . |
Au niveau des hématies, la polymérisation de l'hémoglobine S se traduit par une diminution de la déformabilité, propriété essentielle de cette cellule circulant dans des vaisseaux capillaires de diamètre inférieur au sien. Quand la polymérisation se prolonge, les hématies prennent une forme en faucille. Il s'agit d'un processus de falciformation caractéristique du sang veineux des homozygotes (HbS//HbS).
La polymérisation est un processus coopératif qui demande un certain délai d'initiation. Il y a donc une course de vitesse entre le temps de passage de l'hématie dans ce goulot d'étranglement qu'est le capillaire et le délai de polymérisation qui transforme un globule flexible en une particule rigide et donc susceptible de rester bloqué.
Les interactions entre le génotype, le phénotype et l'environnement
Des variations de la séquence nucléotidique, sans conséquence pathologique directe, sont fréquentes. Elles sont désignées sous le terme de polymorphisme. Lorsque ces modifications portent sur des sites reconnus très spécifiquement par certaines endonucléases (enzymes de restriction) elles sont faciles à mettre en évidence par les méthodes de cartographie génique (polymorphisme de taille des fragments de restriction). L'association de plusieurs de ces polymorphismes définit un haplotype. L'un des résultats les plus intéressants des études des polymorphismes de l'environnement du gène drépanocytaire a été l'observation d'un déséquilibre de liaison : les polymorphismes ne sont pas distribués au hasard mais forment un petit nombre d'haplotypes bien définis. La mutation drépanocytaire a été trouvée habituellement associée à 5 haplotypes désignées d'après leur épicentre. Ce sont les haplotypes Sénégal, Bénin, Cameroun, Bantou et Arabo-indien.
Ils sont des marqueurs d'un environnement chromosomique caractéristique du gène muté, environnement pouvant donner lieu à des particularités génétiques telles que le mode d'expression de l'Hb F. Les enzymes de restriction et les sites utilisés dans la définition des haplotypes drépanocytaires sont représentés dans un schéma.
D'autres hémoglobines sont ainsi en interaction avec l'Hb S à l'intérieur de la cellule. Il existe toujours environ 3% d'Hb A2 et il y a souvent de l'Hb F mais à un taux variable d'un sujet à l'autre et d'une cellule à l'autre. Chez les sujets hétérozygotes l'Hb S est évidemment en interaction avec une autre hémoglobine, normale ou anormale, qui peut faciliter (Hb D Punjab) ou, au contraire, inhiber ( Hb Korle Bu) la polymérisation. Il existe donc une influence génétique large.
Lors d'un effort physique important ou lors d'une exposition de l'organisme à l'altitude, l'hémoglobine se désature en oxygène, on assiste à une falciformation accélérée ce qui indique une intervention de l'environnement sur le phénotype de l'individu atteint de drépanocytose. Toute condition désaturant l'hémoglobine en oxygène est un facteur de risque de falciformation chez les sujets drépanocytaires. Les séjours en altitude sont dangereux et la pratique de sport intensif leur est interdite. Avant tout vol en avion des conseils sont donnés pour éviter des risques d'accidents.
Pourquoi l'allèle muté du gène b a-t-il conféré un avantage sélectif à certaines populations et y devenir plus fréquent au sein de cette population ?
Autrefois la drépanocytose était une anomalie génétique létale responsable d'environ 100 000 morts par an. Environ 80% des homozygotes (HbS//HbS) mourraient alors avant l'âge de la reproduction. Compte-tenu d'une sélection aussi puissante contre le gène HbS, il a été longtemps difficile de comprendre pourquoi, dans certaines populations humaines, sa fréquence atteint et même dépasse largement les 10 %.
Comparant
les cartes de répartition de la malaria d'une part, de la drépanocytose
d'autre part, Haldane fut frappé de leur ressemblance. Une telle
coïncidence suggérait que l'hémoglobine HbS pouvait
apporter un avantage en milieu impaludé. En effet, si l'homozygote
(HbS//HbS) succombe de drépanocytose, l'hétérozygote
(HbA//HbS) résiste mieux au paludisme. Une explication schématique
: lorsque le parasite de la malaria s'installe dans une hématie,
il en détruit l'hémoglobine; l'hématie est mal oxygénée
ce qui provoque des déformations supplémentaires. L'hématie
est alors détruite et avec elle le parasite qu'elle contient. Comme
les hématies non parasitées sont majoritaires, le sujet survit
bien dès lors que ses parasites sont éliminés
régulièrement. Les hétérozygotes (HbS//HbA)
ont donc une probabilité de survie plus grande que les homozygotes
(les homozygotes (HbA//HbA) meurent des suites de la malaria; les homozygotes
(HbS//HbS) ne sont pas protégés).
Le phénotype drépanocytaire résulte de processus biologiques gouvernés par l'expression de plusieurs gènes
Hémoglobines embryonnaires et foetales
Durant la vie embryonnaire, deux types de sous-unités de la famille a sont présentes : la chaîne z appararaît la première, puis la chaîne a.
Il existe également deux chaînes de type b : la chaîne e, spécifique de cette période initiale de la vie, et les chaînes g (ou foetales). Ces diverses sous-unités constituent les trois hémoglobines de l'embryon, l'hémoglobine Gower 1, l'hémoglobine Gower 2 et l'hémoglobine Portland.
L'hémoglobine F, détectable à partir de la 5ème semaine, est le constituant hémoglobinique principal de cette période de la vie. L'hémoglobine F est synthétisée dès les premiers stades de la gestation ; elle atteint entre la 8ème et la 10ème semaine un taux de 90 % qui reste ensuite à peu près constant jusqu'à la naissance. La sous-unité foetale g est en fait constituée par un mélange en proportions variables de deux espèces moléculaires très voisines, produits de deux gènes distincts, les chaînes Ag et Gg qui ne diffèrent que par la nature du résidu en position 136, alanine dans le premier cas, glycocolle dans le second.
Hémoglobines de l'adulte
L'hémoglobine A, représente plus
de 95 % de la totalité des hémoglobines. Il existe en outre
un constituant mineur, l'hémoglobine A2, dont la synthèse
débute dans la période néonatale et qui est exprimée
à un taux d'environ 2,5 %.
Chez l'adulte normal, l'hémoglobine F
ne subsiste plus qu'à l'état de traces inférieures
à 1 % et reste limitée à une population cellulaire
restreinte, les "cellules F". Ces dernières, dont le nombre semble
génétiquement déterminé, représentent
1 à 7 % de l'ensemble des érythrocytes, et correspondraient
à des hématies dont la différenciation est différente
de celle des cellules ne synthétisant que de l'hémoglobine
A.
|
a b g d e t
|
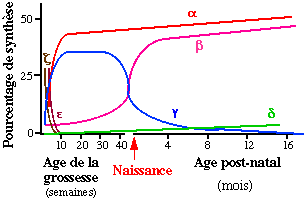
La persistance de l'hémoglobine foetale
après la naissance peut inhiber la falciformation.
Le pourcentage d'hématies contenant de
l'hémoglobine F (cellules F) peut dans certaines cas être
supérieure à 7%.
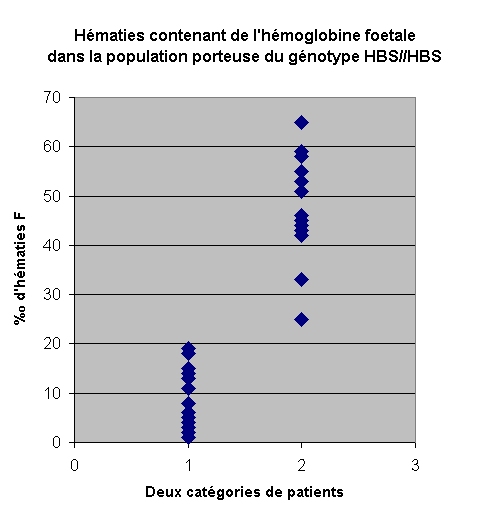
Parmi une population d'individus porteurs du génotype
HBS//HBS on a identifié deux catégories de patients : certains
développent des crises fréquentes et graves (accidents vascullaires)
dues à la falciformation des hématies, d'autres ne développent
qu'exceptionnellement des crises qui sont le plus souvent bénignes.
Les patients qui souffrent de crises caratéristiques
de la drépanocytose ont un taux d'hématies de type F inférieur
à 10%o
Pourquoi la polymérisation de HBS et la falciformation est elle perturbée par la présence d'hémoglobine foetale ? C'est un fait, l'hémoglobine foetale ne s'intègre pas dans le polymère "PolyHBS" portant atteinte à la fois aux contacts verticaux et horizontaux du polymère.
Ainsi même si le phénotype drépanocytaire est monogénique, il est influencé par l'activité d'autres gènes, ici le gène g.
Qu'est-ce qui provoque les crises de la drépanocytose ? Comment vivre avec la drépanocytose ?
Qui dit anémie dit mauvaise tolérance aux efforts physiques puisque la fatigue, induite par l'anémie, est permanente mais elle est aggravée par les efforts. Ces efforts demandent de l'oxygène et ceci entraîne une fatigue plus importante.
Plusieurs facteurs favorisent les crises drépanocytaires :
Tout ce qui désature l'hémoglobine en oxygène déclenche une falciformation accélérée. De nombreux facteurs de l'environnement agissent sur le phénotype d'un individu atteint de drépanocytose.La déshydratation fait perdre de l'eau au globule rouge ce qui rend le sang moins fluide. Or la déshydratation est fréquente chez le drépanocytaire car il est atteint de polyurie due aux lésions provoquées par les petits bouchons de globules rouges. Le rein perd ainsi sa capacité à concentrer les urines. Pour éliminer les déchets, un drépanocytaire est donc obligé d'uriner beaucoup plus qu'un individu non drépanocytaire. Le drépanocytaire doit boire beaucoup. Le ralentissement de la circulation sanguine : Tout ce qui ralentit la circulation peut créer une stase, c'est-à-dire que les globules rouges restent à un endroit et vont favoriser la crise. De nombreuses conditions ralentissent la circulation sanguine : l'effet que l'on appelle garrot (un vêtement trop serré par exemple); une mauvaise position; le froid (contracte les petits vaisseaux et ralentit la circulation); la fièvre (la déshydratation et la formation de protéines inflammatoires ralentissent la circulation); les infections (les globules blancs en excès collent aux vaisseaux et empêchent les globules rouges de circuler). Il faut donc combattre la fièvre avec des médicaments et boire beaucoup. Tout ce qui fait consommer de l'oxygène en plus favorise la crise : Les efforts avec essoufflements, les efforts musculaires concentrés sur un muscle, comme l'haltérophilie, font consommer plus d'oxygène.
La vie en altitude : Le risque est variable d'un patient à l'autre, mais il faut tenir compte qu'au-delà de 1500 m le risque augmente si on n'est pas en condition physique optimale. Mieux vaut éviter les altitudes au-delà de 2000m, contrôler les autres facteurs (froid, neige, efforts physiques). Les voyages en avion : La pressurisation des avions correspond à une altitude de 1500 à 1800 m ce qui constitue un risque certain de crise douloureuses à cause de la baisse d'oxygène. Le passager devra boire abondamment, éviter la station assise prolongée, éviter les vêtements trop serrés. A la mer, à la piscine : attention aux écarts de températures entre l'air et l'eau, sources de crises. Il ne faut pas rester dans l'eau plus de 20 minutes et bien se couvrir (peignoir) en sortant. L'alcool : qui est toxique est contre-indiqué chez les drépanocytaires. L'alcool déshydrate et peut déclencher des crises. Le tabac : est très nocif pour le drépanocytaire dans la mesure où il diminue l'oxygène dans le sang.