|
|
||
Données Activités Observer Comparer Construire Grouper Arbre Glossaire Méthodes
Mises
au point
- Lecture d'un arbre phylogénétique. Notion d'ancêtre commun |
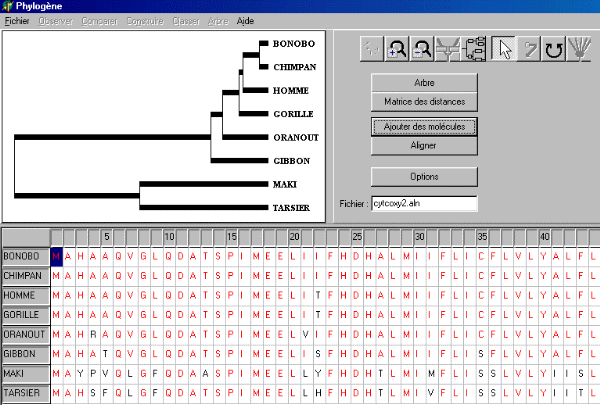
Remarque : plusieurs centaines de milliers de séquences sont maintenant connues, appartenant à des milliers d'organismes. Cependant, pour un organisme particulier, le nombre de gènes séquencés reste limité. Pour certaines collections (les Vertébrés en particulier), il est encore impossible de choisir un ensemble de taxons présents dans Phylogène, d'y travailler avec les données anatomiques et morphologiques, et de poursuivre le travail sur les mêmes taxons avec des données moléculaires. Tracer un arbre à partir de données moléculaires Phylogène permet de tracer un arbre à partir d'un fichier contenant des séquences nucléiques ou protéiques (code universel 1 lettre des acides aminés). Ces séquences peuvent être alignées ou non. A partir d'un fichier de séquences alignées Plusieurs fichiers de séquences alignées sont fournis avec le logiciel. Quelques unes sont adaptées à l'étude de ces relations de parentés sur l'ensemble du vivant. D'autres sont appropriées pour mettre en évidence la notion de famille multigénique ainsi que l'histoire des allèles d'un gène au cours de l'évolution. Ces fichiers au format .aln ont été alignés préalablement avec le logiciel ClustalW. Pour accéder à ces fichiers, choisir dans le menu de Phylogène Fichier, puis Ouvrir un Tableau de molécules. En choisissant le fichier cytoxy2.aln (Collection des Archontes) et en cliquant ensuite sur le bouton "Arbre", on obtient le résultat suivant :
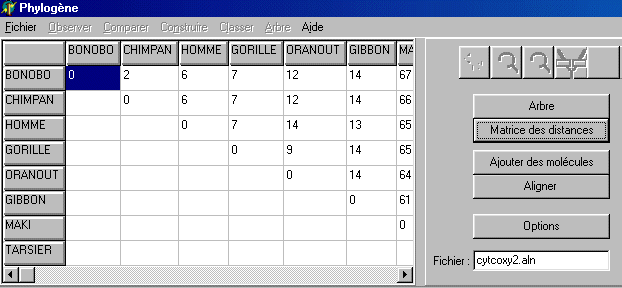
En cliquant sur les boutons portant les noms des taxons, on retire ou on rajoute ces taxons dans l'arbre (si le paramètre "enlever-taxon=oui" dans le fichier phylini.ini. Ce paramètre doit avoir la valeur non si Phylogène est utilisé au collège (Manipulation des taxons et des branches non souhaitable). Matrice des distances La matrice des distances associée à cet arbre est obtenue en cliquant sur le bouton correspondant.
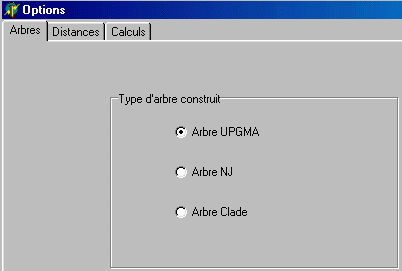
Options Arbres A un fichier de molécule affiché, on peut ajouter une nouvelle séquence. Celle-ci, de même nature que les séquences du fichier affiché, doit être au format PIR avec comme extension .pro ou .adn. L'ajout d'une séquence appelle logiquement un réalignement de l'ensemble des séquences. A manipuler donc avec prudence. A partir d 'un fichier de séquences non alignées Phylogène (comme Clustal) accepte d'aligner des séquences contenues dans un seul fichier au format PIR. Dans ce format, chaque séquence est précédée d'une ligne comportant le signe > suivi du nom de la séquence (en un seul mot). La séquence ne doit comporter aucun espacement. Il n'y pas non plus de ligne vide entre deux séquences. Exemple :
|
|