
5 - Arbre phylogénétique des génomes séquencés de fin décembre à fin février à l’échelle mondiale
5 - Arbre phylogénétique des génomes séquencés de fin décembre à fin février à l’échelle mondiale
Cliquer sur l'image pour accéder aux données sur Nextstrain.
Légende
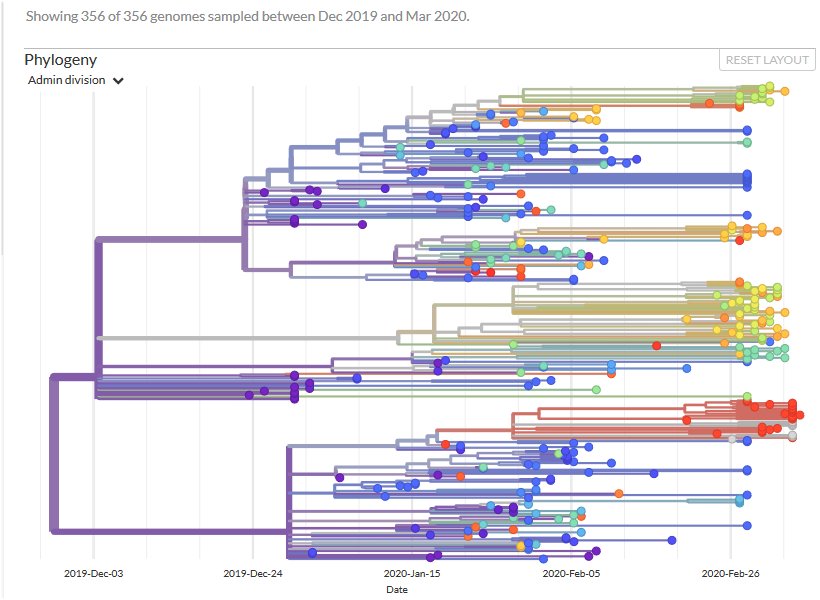
Chaque cercle coloré correspond à un génome viral réellement séquencé. La couleur des ronds renseigne sur le pays où a été prélevé le virus (Dans Nextstrain, cliquer sur le petit triangle de « admin divison » pour voir le code couleur de chaque pays). Ainsi les ronds violets indiquent que le virus a été prélevé chez des personnes chinoises. Les dates « sur l’axe des X » permettent de situer approximativement celle où le génome viral a été recueilli. Les branches renseignent sur les relations de parenté entre les génomes viraux reconstitués à partir des algorithmes utilisés par Nextstrain.
Quand on clique soit sur une branche soit sur un « rond », un rectangle noir s'affiche qui renseigne sur les mutations ayant eu lieu. On n’indique que les mutations spécifiques à cet endroit de l’arbre phylogénétique.
Exploitation pédagogique
Les élèves doivent utiliser les principes de construction des arbres envisagés au point 4 et les informations fournies par les rectangles. Ces informations indiquent le pays où le génome séquencé a été trouvé, la nature de l’évolution du génome au niveau nucléique et au niveau protéique (cela par rapport au génome de référence). La numérotation de l’évolution nucléique est faite par rapport au génome total du virus qui comprend un peu plus de 29000 nucléotides. Celle de l’évolution protéique renseigne sur la protéine précise dont la séquence est modifiée. La nature des acides aminés est fournie dans le code à une lettre.
Outre les relations de parenté fondées sur le partage de mutations communes, les arbres de Nexstrain renseignent sur la date de séquençage du virus chez chaque individu.
On peut leur demander :
- De situer la racine de l’arbre, d’indiquer en quoi cet arbre révèle que le virus est apparu en Chine puis s’est répandu dans diverses régions du monde, de rechercher s’il y a eu dès le mois de janvier une diversification du génome du virus dans la population chinoise.
La racine de l’arbre se trouve bien sûr à gauche. Lorsqu’on clique sur les lignes verticales de la racine, on constate qu’il est indiqué 326 descendants et aucune mutation nucléique. Cela indique que le Sars-Cov-2 a une origine unique et que l’arbre traduit les relations de parenté entre les virus séquencés de janvier à fin février 2020. La couleur violette des cercles représentants des virus séquencés en janvier et des branches qui les relient indique que ces virus avaient infecté des chinois. En février, les couleurs des cercles des virus séquencés sont plus diversifiées ce qui indique qu’ils proviennent de différentes régions du monde notamment d’Asie et des USA. Cela traduit la propagation du virus dans la planète à partir de la Chine.
En cliquant sur les cercles violets représentatifs des génomes du virus en Chine en janvier, on constate que pour certains l’arbre n'indique aucune mutation nucléique, donc un génome identique à celui du virus de référence, publié au tout début janvier et qui était issu d’une personne victime de la Covid de Wuhan (Chine). En revanche les informations relatives à d’autres virus (cercles violets) indiquent 1 à 4 mutations nucléiques. Cela traduit une évolution par mutations ponctuelles de la population de virus en Chine dès le début de l’infection. Ces mutations sont des substitutions de nucléotides.
En cliquant sur la branche horizontale du bas de l’arbre, on constate deux mutations nucléiques C8782T et T28144C et une seule mutation protéique affectant la protéine ORF8 du virus : L84S.
Cela s’explique par la redondance du code génétique : la mutation nucléique T28144C n’entraîne aucun changement dans la séquence de la protéine ORF8.
L'Arbre légendé présente l’intérêt de fournir une vue d’ensemble des caractéristiques des génomes de plusieurs virus séquencés à divers moments entre janvier et mars. Il illustre une diversification par accumulation de mutations successives différentes dans les divers virus. Certains ont le même génome que le génome de référence de Wuhan alors que d’autres possèdent de une à cinq mutations .
{kind=link}
- Considérer l’arbre mettant en évidence un sous clone dans l’arbre précédemment analysé.
Cliquer sur l'image pour accéder aux données sur Nextstrain.
- Montrer en quoi les informations fournies illustrent ces deux connaissances du programme : toute mutation devient pérenne pour toute la lignée qui dérive du mutant ; la diversité génétique dans un clone résulte de l’accumulation de mutations successives.
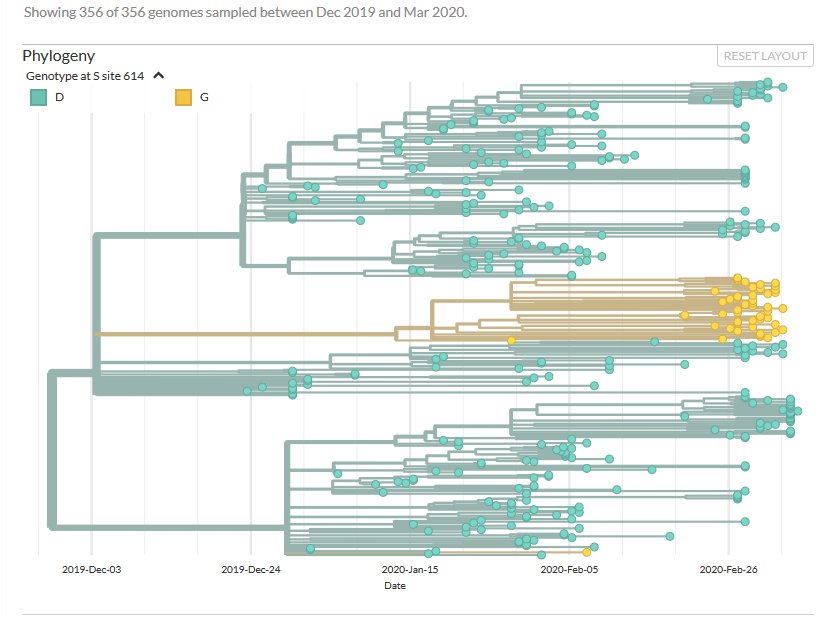
La coloration de cet arbre est basée sur le gène codant pour la protéine « spike » S qui se lie au récepteur ACE2 présent sur les cellules cibles et permet l’entrée du virus dans ces cellules. Deux allèles sont présents dans la population de virus séquencés. L’un au site 614 de la protéine code pour l’acide aminé D (acide aspartique) en vert, l’autre pour l’acide aminé G (glycine) en jaune. La couleur uniquement verte des virus séquencés au tout début de l’épidémie indique que l’allèle ancestral est celui codant pour l’acide aminé D. L’allèle G résulte donc d’une mutation ponctuelle : D614G.
En cliquant sur la branche jaune à ses débuts, on obtient des informations complémentaires : il y a eu 3 mutations nucléiques : C241T ; C3037T ; A23403G caractérisant le mutant. Deux d’entre elles ne modifient pas la séquence de la protéine S ; la troisième A23403G est à l’origine du mutant protéique 614G.
L’arbre indique que tous les virus de couleur jaune sont plus étroitement apparentés entre eux qu’avec les autres virus séquencés. Ils possèdent tous la mutation D614G et dérivent du premier mutant ayant acquis cette mutation. Au sein du clone de 326 génomes, ils forment un sous-clone de 48 virus défini par cette mutation.
Néanmoins au sein de ce sous clone, les virus n’ont pas tous exactement le même génome. En cliquant sur les divers virus de fin février, on constate que plusieurs possèdent de nouvelles mutations. Cela est en accord avec la notion du programme que la diversité génétique dans un clone résulte de l’accumulation de mutations successives.
L’arbre sur les sous clones traduit l’ensemble des sous clones du Sars-Cov-2 identifiés au terme de 2 mois de l’évolution de la population de virus.
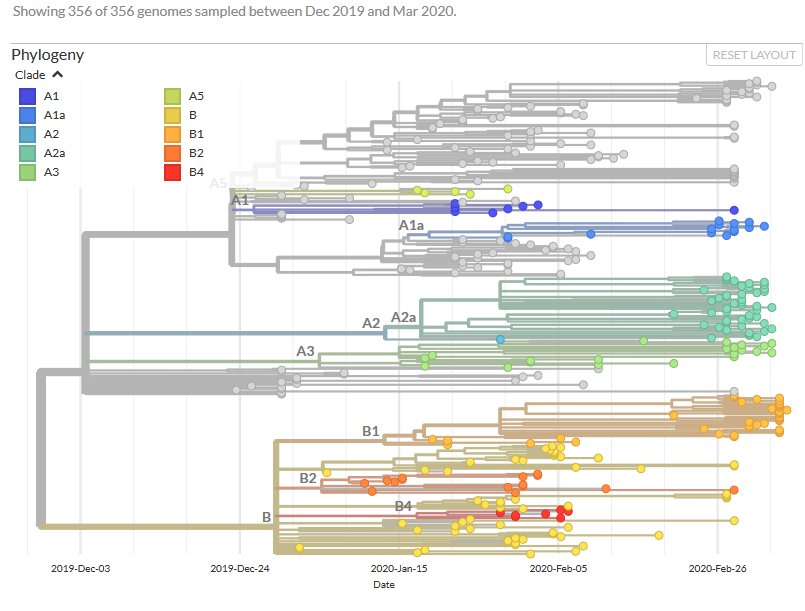
- On peut demander aux élèves d’indiquer la référence du clone défini par la mutation D614G, d’indiquer ce qui défini le sous clone A2a, le sous clone B et d’autres sous clones si on le souhaite.
Cliquer sur l'image pour accéder aux données sur Nextstrain.
En cliquant sur les divers sous clones, on constate que seul le sous clone A2 est défini par la mutation protéique S : D614G.
En cliquant sur A2a , on trouve que ce sous clone est défini par la mutation nucléique C14408T et la mutation protéique dans ORF1b : P314L.
Tous les virus de A2a possèdent aussi la mutation D614G puisque A2a est un sous clone de A2.
Le sous clone B est défini par les mutations nucléiques C8782T, T28144C et la mutation protéique ORF8 : L84S
On peut constater que fin-février - début mars, le nombre de virus séquencés est déjà important et qu’ils appartiennent à plusieurs sous clones différents ce qui traduit l’importance de la diversification des génomes de Sars-Cov-2 en deux mois.