
Arbre phylogénétique des génomes séquencés de fin décembre à fin février à l’échelle mondiale
6 . Arbre phylogénétique des génomes séquencés de fin décembre à fin février à l’échelle mondiale
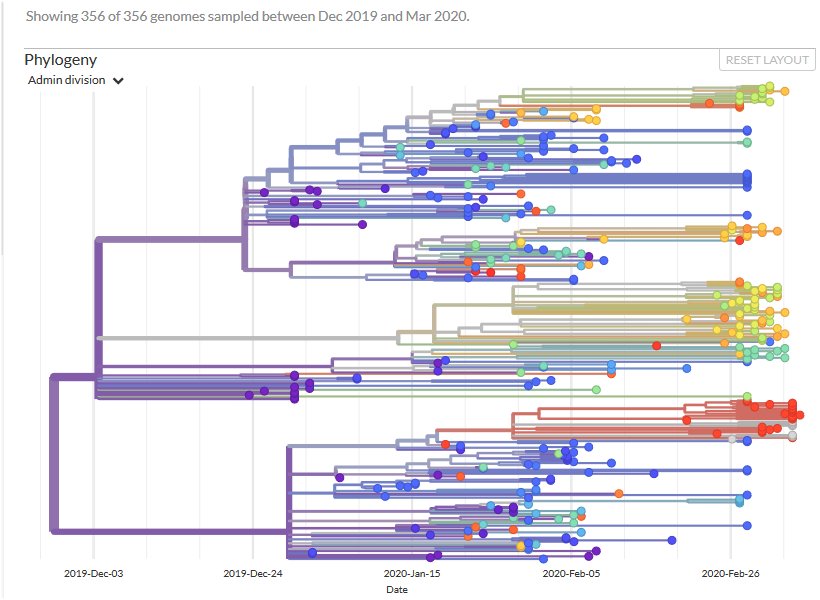
Cliquer sur l'image pour accéder aux données sur Nextstrain.
Légende
Chaque cercle coloré correspond à un génome viral réellement séquencé. La couleur des ronds renseigne sur le pays où a été prélevé le virus (Dans Nextstrain, cliquer sur le petit triangle de « admin divison » pour voir le code couleur de chaque pays). Ainsi les ronds violets indiquent que le virus a été prélevé chez des personnes chinoises. Les dates « sur l’axe des X » permettent de situer approximativement celle où le génome viral a été recueilli. Les branches renseignent sur les relations de parenté entre les génomes viraux reconstitués à partir des algorithmes utilisés par Nextstrain.
Quand on clique soit sur une branche soit sur un « rond », un rectangle noir s'affiche qui renseigne sur les mutations ayant eu lieu. On n’indique que les mutations spécifiques à cet endroit de l’arbre phylogénétique.
{kind=link}
Exploitation
On peut demander :
- De situer la racine de l’arbre et de dégager des idées générales sur la propagation du virus.
La racine de l’arbre se trouve bien sûr à gauche. La couleur des ronds et celle des branches indiquent que le virus est apparu en Chine. On note que les premiers virus séquencés sont de fin décembre-début janvier et que la racine (branche verticale de gauche) est située en novembre. C’est le traitement des données, notamment la détermination de la vitesse d’évolution du virus, qui conduit les spécialistes à estimer que sa date d’apparition précède de 2 mois environ les premiers cas de patients atteints de la Covid-19. La couleur des ronds vers la fin février traduit l’exportation du virus hors d’Asie et donc l’amorce de la pandémie.
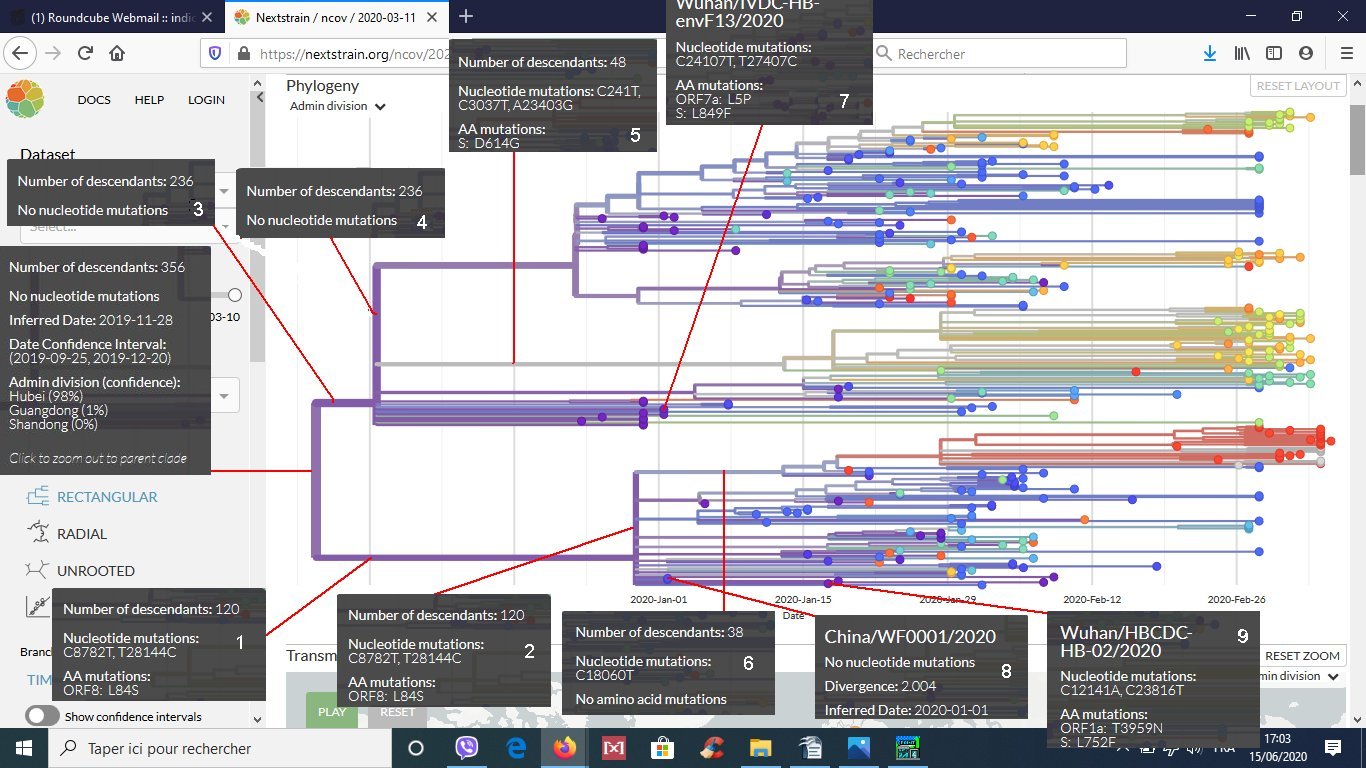
- D’indiquer ce que signifie : nombre de descendants 356
Les 356 génomes séquencés dérivent d’un génome ancestral commun qui est à la racine de l’arbre et qui sert donc de référence. C’est ainsi la phylogénie au sein d’un clone (ou clade) de 356 virus qui est représentée. - D’indiquer les caractéristiques du premier sous clone qui s’est individualisé au cours de l’évolution du Sars-CoV-2 (sous clone défini par les deux mutations nucléiques : C8782T et T28144C).
- De proposer une explication au fait que ce sous clone est caractérisé par deux mutations nucléiques et une seule mutation protéique (les mutations sont des substitutions et par suite du caractère redondant du code génétique, une substitution au niveau nucléique peut ne pas entraîner une substitution au niveau protéique).
- D’indiquer ce que montre le rectangle 6 (c’est un sous clone de 38 virus du sous clone de 120 virus caractérisé par une mutation nucléique sans effet au niveau protéique).
- D’indiquer les caractéristiques des génomes ancestraux indiquées sur le rectangle 4 (aucune mutation n’est intervenue à partir de la racine et par conséquent tous ces virus ont la même séquence que celle de référence).
- D’expliquer pourquoi sur le rectangle 4 il est indiqué : 236 descendants et sur le rectangle 5, 48 descendants (le rectangle 5 se rapporte à un sous clone du clone de 356 défini par 3 mutations nucléiques et une seule mutation protéique : S : D614G).
- D’indiquer si les virus recueillis à Wuhan (rectangles 7 et 9 ) font partie d’un même sous clone (non, car ils ne possèdent pas les mêmes mutations).
- D’indiquer par rapport à la séquence de référence les caractéristiques du génome du virus de Wuhan (rectangle 9) au niveau nucléique (C8782T ; T28144C ; C12141A ; C23816T) et protéique ( ORF8 : L84S ; ORF1a : T3959N ; S : L 752 F.
Remarques : La numérotation du nucléotide muté est faite par rapport au génome total du virus, qui comprend un peu plus de 29000 nucléotides. La numérotation de l’acide aminé muté se fait par rapport à une protéine précise du virus (Sars-CoV-2 en possède 26).
Considérons la mutation au niveau protéique S : D614G. S désigne la protéine « spike » (celle qui permet au virus de s’attacher au récepteur cellulaire ACE2 des cellules qu'il va parasiter). La mutation dans la séquence de la protéine S est située au 614ème acide aminé et consiste en la substitution d’une glycine (G) à l’acide aspartique (D). La mutation correspondante au niveau nucléique est A23403G, qui ne se réfère pas au gène codant pour la protéine S mais au génome total.