Exploitation pédagogique
Exploitation pédagogique du DICS-X
Les intentions de ce dossier
La documentation de ce dossier sur le DICS-X permet :
- De traiter un exemple d’hérédité lié au sexe, notion propre à la classe de terminale par rapport à la classe de première.
- De réinvestir et donc renforcer les notions de phénotype clinique, de phénotype cellulaire et de phénotype moléculaire ainsi que les relations entre ces différents niveaux de phénotype, ce qui implique d’utiliser les notions de base d’immunologie vue en première.
- De réinvestir les raisonnements permettant de faire une hypothèse sur la localisation chromosomique d’une gène à partir des données phénotypiques d’un arbre généalogique et de la tester à partir des données moléculaires sur le génotype de plusieurs individus d’une famille.
- De réinvestir les connaissances sur le brassage génétique pour discuter de la compatibilité HLA des membres d’une famille, en particulier des frères et soeurs.
- D’introduire, lors de la mise en évidence du gène en cause, la notion de cellule souche hématopoïétique, notion indispensable pour la compréhension des thérapies utilisées dans cette maladie héréditaire et pour d’autres maladies comme les hémoglobinopathies.
- D’aborder, en s’appuyant sur les connaissances acquises sur les mécanismes cellulaires et moléculaires du DICS-X, le principe des protocoles de thérapies susceptibles d’aboutir à une guérison des patients. C’est sans doute un des exemples les plus parlants pour montrer le lien entre la recherche fondamentale et les applications médicales.
1 - Les mécanismes cellulaires et moléculaires du DICS-X
a - Phénotypes cliniques et cellulaires du DICS-X, maladie héréditaire liée au sexe (doc 1)
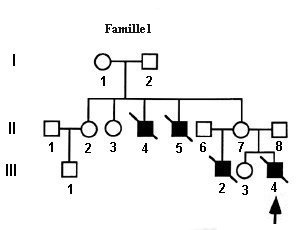
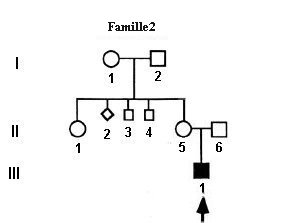
Les arbres généalogiques des trois familles établis initialement à partir du phénotype clinique, permettent de renforcer les raisonnements d’analyse génétique. Le fait que les seuls membres atteints dans les trois familles soient des garçons, conduit à l’hypothèse que le DISC-X est une maladie liée au sexe où le gène en cause est situé sur le chromosome X.
 |
 |
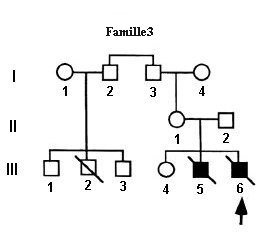 |
On peut renforcer cette hypothèse en demandant aux élèves de se placer dans l’hypothèse inverse d’hérédité autosomale. Dans ce cas, les hommes initialement à l’extérieur de la famille, qui ont comme conjoint une femme de la famille et un fils malade, doivent être hétérozygotes. Cela est très peu probable vu que c’est une maladie rare (1/100.000 naissances). Le cas de la femme qui a eu deux garçons « DICS-X » ayant des pères différents est particulièrement informatif. On arrive à la conclusion que dans les familles, la maladie se transmet par l’intermédiaire des femmes hétérozygotes, chez lesquelles le phénotype « DICS-X » est dominant.
Les élèves doivent arriver à l’idée que, puisque le phénotype clinique traduit une déficience immunitaire, les phénotypes cellulaires doivent être relatifs à des cellules du système immunitaire, ce que permet de tester le document. Ils arrivent ainsi à la conclusion que la maladie est due au nombre très faible ou nul des lymphocytes T (T4 et T8), cellules de l’immunité adaptative, et des cellules NK, cellules de l’immunité innée.
b - Le gène IL2RG et ses allèles mutés dont l’expression est à l’origine de la maladie
Il s’agit maintenant d’identifier le gène qui, en mutant, est à l’origine de cette déficience des cellules immunitaires. Pour cela le document 2 fait le point sur les mécanismes du développement des lymphocytes à partir des cellules souches hématopoïétiques (CSH) de la moelle osseuse, notion centrale pour la suite.
Le document « précisions sur le développement des lymphocytes » apporte l’information essentielle, à savoir que le développement des précurseurs (on utilise aussi le terme de progéniteurs) des lymphocytes dans la moelle osseuse, donc à un stade précoce, est dépendante d’une protéine appelée gamma c. Celle-ci est indispensable à la multiplication des précurseurs qui est très importante.
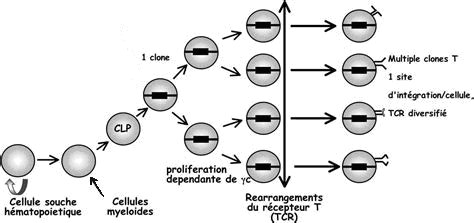
Le document sur les interleukines permet de préciser le rôle de cette protéine gamma c. C’est un constituant commun à plusieurs récepteurs aux interleukines présents sur la membrane des précurseurs des lymphocytes T. Sur le schéma, la couleur bleue visualise la présence de la protéine gamma c dans les récepteurs à des interleukines différents. Les élèves doivent mobiliser leurs connaissances sur le mécanisme de réponse des cellules aux messages chimiques qu’elles reçoivent, pour saisir qu’en l’absence de cette protéine, les précurseurs lymphocytaires ne peuvent répondre aux messages qui leur sont délivrés dans la moelle osseuse, et donc proliférer.
Le gène codant pour la protéine gamma c est désigné par IL2RG. Les élèves disposent de deux fichiers de séquences. L’un porte sur les séquences codantes de l’allèle « sain » de référence et de plusieurs allèles trouvés chez des garçons malades. L’autre sur les génotypes de membres des familles précédemment envisagées.
Fichier : « Allèles du gène IL2RG.edi »
Fichiers « DICS-X-famille1-Bis.edi », « DICS-X-famille2-Bis.edi », « DICS-X-famille3-Bis.edi »
Le fichier sur les génotypes indique que les garçons n’ont qu’un seul allèle du gène et les filles deux. Cela confirme l’hérédité liée au sexe. Il confirme aussi que les mères des garçons malades sont hétérozygotes (sauf dans la famille 2) : elles possèdent le même allèle muté que celui trouvé chez leur fils.
Remarque sur le phénotype des femmes hétérozygotes.
On sait que chez les femmes un des deux chromosomes X est Inactivé. Si le chromosome portant l’allèle normal est inactivé, on devrait s’attendre à ce que la femme présente des symptômes du DICS-X. Ce n’est pas le cas. Des recherches ont montré que chez les femmes hétérozygotes, c’est toujours le chromosome portant l’allèle muté qui est inactivé.
La famille 2 révèle un mécanisme différent de celui des deux autres familles. La mère et la grand-mère du garçon malade sont homozygotes avec deux allèles « sains ».
L’allèle muté présent chez le garçon ne peut résulter que d’une mutation de novo probablement au cours de l’ovogénèse de sa mère à l’origine de l’ovocyte qui après fécondation lui a donné naissance.
Fichier des séquences des allèles du gène IL2RG
Allèles du gène IL2RG de la famille-Bis.edi (Mère et enfant).
L’exploitation de ces séquences avec Anagène ou Genigen 2 doit conduire les élèves au constat suivant (les indications relatives à un allèle muté le sont par rapport à l’allèle de référence).
| Allèle | Nucléotide muté | Codon muté | Impact sur la protéine gamma c |
| Allèle 1 | T186A | TGT62TGA | Protéine réduite à 62 acides aminés sur 369 |
| Allèle 2 | G141 | GGC114GAC | Gly114Asp |
| Allèle 3 | T373A | TAC 126 AAC | Tyr126Asn |
| Allèle 4 | C430T | CAG144TAG | Protéine réduite à 143 acides aminés sur 369 |
| Allèle 5 | T458A | ATC153AAC | Ile153Asn |
| Allèle 6 | 126 del G | CTG42CTA | Protéine ne possédant que les 42 premiers acides aminés communs avec la protéine de référence. Après, décalage du cadre de lecture. |
| Allèle 7 | 353 ins A | La protéine mutée a seulement les 120 premiers acides aminés en commun avec la protéine de référence. |
En ce qui concerne le septième allèle, l’insertion d’un nucléotide A se fait dans un groupe de 7 nucléotides A. Le logiciel arbitrairement le place en première position de ce groupe qui comprend chez l’allèle muté 8 nucléotides A de suite. L’article des chercheurs le place à la fin. Cela n’a pas grande importance.
On peut demander aux élèves de tirer les conclusions principales d’un tel tableau.
Les mutations ayant abouti à ces allèles sont des substitutions non sens et faux sens, des insertions et délétions. Cela renforce la notion de poly-allélisme des gènes à l’origine des maladies héréditaires. Surtout, cela amène à penser que dans cette maladie comme dans d’autres, les conséquences phénotypiques des allèles mutés sont différentes. Par exemple, les allèles codant pour des protéines très courtes par rapport à la protéine de référence (comme la protéine codée par l’allèle 1) sont peu stables et se dégradent. Les précurseurs de lymphocytes dans ces cas là n’ont pas de protéine gamma c membranaire et sont incapables de se multiplier. Cela conduit à un phénotype clinique sévère. Lorsque la protéine est beaucoup plus longue, bien que moins fonctionnelle que la protéine saine, elle peut persister dans les récepteurs membranaires des précurseurs et conduire à un phénotype clinique moins sévère.
2 - Les traitements thérapeutiques du DICS-X (Documents 3, 4 et 5)
a - Les informations fournies par la greffe de moelle osseuse
Les traitements visant à limiter le risque d’infections reposent sur l’utilisation d’antibiotiques, d‘immunoglobulines, et le placement en milieu stérile des jeunes enfants. Le seul traitement curatif, c’est-à-dire visant à restaurer un système immunitaire fonctionnel avec des lymphocytes T, est la transplantation de CSH d’un donneur en bonne santé. Cette greffe est précédée d’une période dite de « conditionnement » pour détruire les cellules de la moelle osseuse du patient, et permettre que leurs places soient prises par les cellules du receveur.
Pratiquée pour la première fois en 1968, cette greffe allogénique est confrontée au problème de la compatibilité génétique entre les systèmes HLA du donneur et du patient receveur. Le cas le plus favorable est celui où les deux systèmes HLA sont identiques. Dans ce cas, les cellules du système immunitaire reconstitué du patient à partir des CSH du donneur ne vont pas engendrer des réactions immunitaires contre ses propres organes. Ce cas de compatibilité génétique peut se trouver dans un cadre familial lorsque le patient a une soeur ou un frère ayant le même système HLA. Dans le cas de la famille du document 3, c’est l’enfant 2 qui est le plus approprié pour fournir ses CSH à son frère malade. Le calcul de la probabilité pour qu’un frère ou une soeur ait un système HLA identique amène à réinvestir les notions de brassage génétique, ici du brassage inter chromosomique. La probabilité est de ¼. Les élèves doivent saisir que le brassage intra chromosomique est exceptionnellement impliqué car les gènes du système HLA sont étroitement liés.
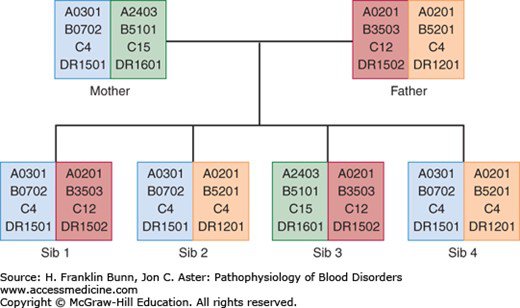
Si le patient n’a pas de frère ou soeur génétiquement compatible, on peut injecter des CSH de sa mère ou de son père qui sont haplo identiques. Cela signifie qu’elles ont un de leur deux chromosomes 6 possédant les mêmes allèles des gènes HLA que le patient. On peut aussi injecter des CSH d’une personne non apparentée mais à condition que son système HLA soit le plus proche possible de celui du patient. Dans tous les cas, si la compatibilité n’est pas totale, il faut suivre attentivement l’évolution du système immunitaire du patient et mettre en place un traitement immunosuppresseur adapté.
Le graphique sur la survie après la transplantation indique qu’elle est supérieure en cas de compatibilité immunologique et ce jusqu’au moins 20 années après.
Cela a montré aux médecins que les CSH injectées pouvaient occuper la moelle osseuse d’un patient DICS-X, et se mettre à fonctionner durablement en produisant les différentes cellules du système immunitaire et notamment les lymphocytes T.
b - Les informations fournies par un cas de thérapie génique dit naturelle
C’est un cas qui a intrigué les médecins et les a conduits à penser qu’un protocole de thérapie génique à partir de CSH du patient modifiées génétiquement pourrait être un traitement curatif, en remplacement de la greffe de moelle osseuse, lorsque celle-ci est rendue impossible par la difficulté à trouver un donneur compatible.
Ce cas est celui d’un garçon affecté par les symptômes du DICS-X et la maladie a été confirmée par un test génétique. A l’âge de un an, il est présenté aux médecins de l’hôpital Necker à Paris. Ceux-ci constatent que les symptômes de la maladie, et notamment les infections, sont moins accusées. A leur surprise, l’analyse sanguine révèle la présence de lymphocytes en nombre un peu sous la normale.
Ils décident alors de faire l’analyse des allèles du gène IL2RG présents dans les lymphocytes T et les polynucléaires du patient ainsi que dans les cellules de sa mère. Le fichier de séquences traité par un logiciel permet d’identifier les résultats obtenus. Les lymphocytes du garçon possèdent l’allèle fonctionnel de référence alors que les polynucléaires (et les autres cellules de l’organisme) possèdent l’allèle muté (TGT115CGT) qui code pour la protéine mutée (Cys115Arg). Le génotype de la mère du patient est constitué par l’allèle muté (TGT115CGT). Il est très probable que l’oncle et le grand oncle du patient décédés l’âge de 4 et 6 mois possédaient aussi cet allèle muté.
Les médecins ont interprété ces constats de la façon suivante. Initialement le garçon a reçu l’allèle muté de sa mère, ce qui explique les symptômes du DICS-X durant sa première année. Le fait qu’à un an ses lymphocytes possèdent l’allèle fonctionnel ne peut s’expliquer que par une mutation « reverse » survenue chez un précurseur des lymphocytes dans la moelle osseuse. La mutation étant un évènement dont la fréquence est faible, les médecins estiment qu’une seule mutation survenue chez un précurseur est à l’origine de lymphocytes T du garçon.
Un seul précurseur est à l’origine d’une grande diversité de lymphocytes T (1000 types).
Ainsi, une forme de thérapie génique « naturelle » avait en partie corrigé le DICS-X du garçon. Cette observation montrait aussi que les précurseurs non mutés avaient un avantage sélectif sur ceux mutés. Surtout, elle a renforcé les médecins dans l’idée qu’il faudrait modifier génétiquement une population de CSH du patient sans qu’elle soit considérable pour constituer un traitement curatif du DICS-X.
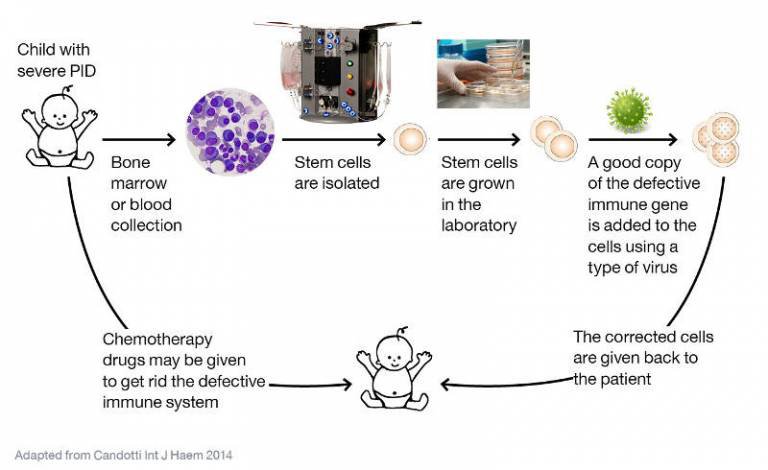
3 - Vue d’ensemble de la première thérapie génique du DICS-X
La transplantation de cellules souches hématopoïétiques, l’observation du cas de thérapie génique naturelle, ont conduit les médecins de Necker à concevoir le protocole de thérapie génique illustré par les deux figures.
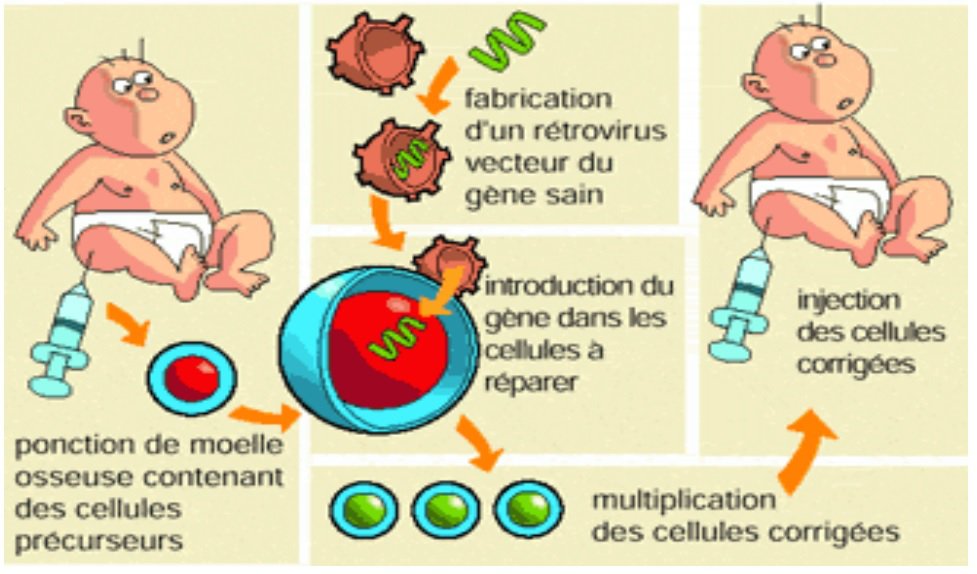
Les élèves doivent repérer que par rapport à la greffe de CSH d’un donneur, il s’agit de transférer des CSH modifiées du patient. Le système HLA des CSH transférées est le même que celui des cellules de l’organisme du patient. Le problème de la compatibilité génétique ne se pose pas.
La modification du génome de CSH du patient réalisée ex-vivo réside dans l’introduction dans ces cellules d’un allèle fonctionnel du gène IL2RG. C’est donc une modification par addition d’un gène qu’on peut qualifier de thérapeutique.
Ce gène est introduit dans les cellules par l’intermédiaire d’un vecteur viral. Il ne s’agit donc pas dans ce premier essai clinique de correction de l’allèle déficient comme dans la thérapie naturelle mais d’addition de l’allèle « sain ».
Les élèves doivent identifier dans les deux figures trois grandes étapes : le recueil de CSH du patient, la modification génétique des CSH prélevées réalisée ex-vivo, la réintroduction dans l’organismes des CSH modifiées.
La deuxième figure indique en plus que le patient est soumis à une chimiothérapie qui détruit ces CSH défectueuses. Cela facilite par la suite l’occupation des CSH thérapeutiques dans la moelle osseuse du patient, dans un environnement favorable.