
Hérédité des rétinites pigmentaires
I . Objectifs et intérêt de l’étude
L’exploitation du dossier sur les myopathies en terminale est à privilégier dans la mesure où il complète l’étude, amorcée en première, des raisonnements à tenir dans l’analyse des arbres généalogiques, en particulier pour tout ce qui concerne l’hérédité liée au sexe. Il permet aussi d’enrichir la notion de diversité des mutations pouvant être impliquées dans une maladie génétique et la notion de néomutation (mutation de novo). Il permet de percevoir comment des mutations d’un même gène peuvent engendrer des phénotypes différents (myopathie de Duchenne et myopathie de Becker). Enfin le dossier montre tout l’intérêt du diagnostic moléculaire.dans l’évaluation du risque génétique.
Le dossier sur la génétique des rétinites pigmentaires a pour objectif de fournir un outil permettant de voir si les élèves sont capables d’appliquer à l’analyse d’une autre maladie génétique, les raisonnements appris avec l’analyse des myopathies.
Il repose sur le fait que les rétinites sont des maladies génétiques pouvant être causées par des mutations de plusieurs gènes, lesquels s’expriment dans les cellules photoréceptrices, notamment les bâtonnets. Les protéines codées par ces gènes ont des actions différentes, mais leurs mutations provoquent à plus ou moins long terme une dégénérescence de ces cellules et donc une cécité plus ou moins accusée.
La majorité de ces gènes sont situés sur des autosomes, mais 15% des rétinites environ sont dues à un gène situé sur le chromosome X.
En outre, le phénotype « rétinite » peut être dominant ou récessif suivant le gène en jeu. Pour le gène de la rhodopsine, la majorité des allèles mutés engendre un phénotype « rétinite » dominant, mais certaines mutations sont cause d’un phénotype récessif de la rétinite.
Ce sont ces caractéristiques qui sont exploitées dans les documents soumis à l’analyse des élèves. Ceux-ci comportent :
- Une présentation des rétinites pigmentaires.
- Des informations sur les gènes dont les mutations peuvent causer des rétinites, notamment leur localisation chromosomique et la dominance ou la récessivité des rétinites engendrées par leurs mutations.
- Des arbres généalogiques relatifs à des familles où certaines personnes sont victimes de rétinites.
- Des séquences des allèles possédés par quelques individus de chaque famille relatifs à au moins deux gènes dans 3 familles sur 4 dont les mutations pourraient être à l’origine de la rétinite dans la famille.
Le travail proposé aux élèves se déroule en trois temps :
- En premier, ils doivent analyser chaque arbre généalogique pour déterminer si le phénotype rétinite est dominant ou récessif et pour savoir si le gène en cause est situé sur un autosome ou un chromosome sexuel. L’analyse ne débouche pas obligatoirement sur une conclusion univoque. Pour plusieurs des arbres proposés, deux interprétations des données sont possibles.
- En second, il faut confronter les conclusions tirées de l’arbre généalogique avec les informations fournies sur les gènes dont les mutations sont à l’origine des rétinites. On doit arriver à faire une hypothèse sur le ou les gènes qui peuvent être mutés dans la famille.
- Enfin, on teste cette hypothèse en exploitant les séquences fournies sur les génotypes de plusieurs personnes de chaque famille.
Le dossier comprend 4 arbres généalogiques. Il est possible de tous les envisager, en partageant éventuellement le travail. Ces arbres sont regroupés deux à deux. Pour chaque groupe de deux arbres, il est proposé de réaliser un calcul de risque génétique faisant intervenir les deux familles, donc deux gènes.
II. Les documents pour les élèves
A - Quelques indications sur le phénotype « rétinite pigmentaire »
Maladies héréditaires, les rétinites pigmentaires sont la cause principale des malvoyances. La maladie débute généralement par une atteinte de la vision nocturne avec réduction du champ visuel causée par la dégénérescence progressive des bâtonnets. La dégénérescence de la rétine s’accoît progressivement et finit par atteindre la région de la fovéa, ce qui entraîne une baisse de l’acuité visuelle et une cécité plus ou moins accusée.
B - Quelques gènes impliqués dans les rétinites pigmentaires
Le phénotype « rétinite pigmentaire » est très hétérogène sur le plan clinique, se traduisant par une variabilité de la précocité des premiers signes et de la durée d’extension de la maladie. Une première raison est due au fait que des mutations de nombreux gènes peuvent être à l’origine de cette maladie génétique. Une deuxième raison réside dans la diversité des mutations pour un même gène
Les principaux gènes impliqués sont les suivants :
- Le gène RHO qui code pour un pigment visuel présent dans les bâtonnets, la rhodopsine. Ce gène est situé sur le chromosome 3. Le phénotype « rétinite » causé par des allèles mutés de ce gène est généralement dominant. Cependant, certaines mutations de ce gène sont à l’origine d’un phénotype « rétinite » récessif.
- Le gène RPGR code pour une protéine des cellules photoréceptrices, cônes et bâtonnets. Il est localisé sur une région propre au chromosome X. Les mutations de ce gène sont à l’origine d’une « rétinite » récessive.
- Le gène PDE code pour une enzyme, la phosphodiestérase qui, dans les cellules photoréceptrices, intervient dans la conversion des photons absorbés par les pigments rétiniens (rhodopsine et pigments des cônes) en un signal nerveux. Ce gène est situé sur le chromosome 4. Le phénotype « rétinite » dû à des allèles mutés de ce gène est récessif.
- Le gène RDS codant pour la périphérine, protéine des cellules photoréceptrices, bâtonnets et cônes, indispensable au maintien de leur structure. Le gène RDS est situé sur le chromosome 6. Le phénotype « rétinite » dû à des mutations de ce gène est dominant.
C. Analyse généalogique et analyse génotypique.
a - Hérédité autosomale- Hérédité liée au sexe
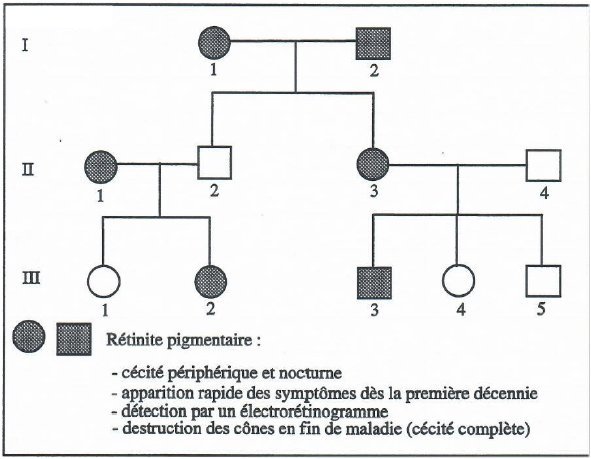
- Arbre généalogique de la famille Pierre
Génotypes famille Pierre Rhodopsine.edi
Génotypes famille Pierre Périphérine.edi
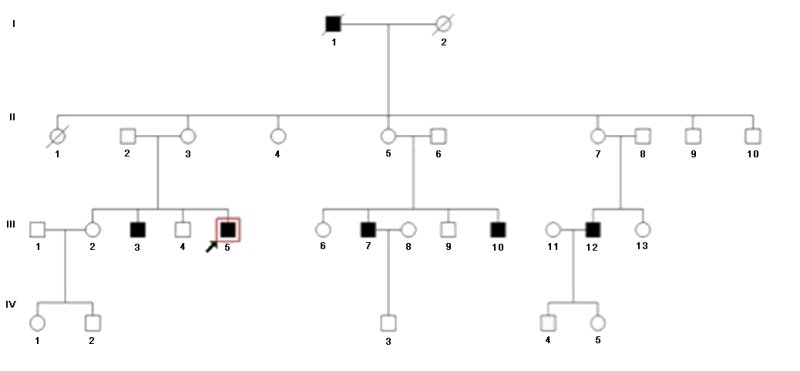
- Arbre généalogique de la famille Paul
Source : Z Zang et al. Novel mutations of RPGR in chinese families with X-linked retinitis pigmentosa. BMC Ophtalmol. 2019 Nov 27
Génotypes famille Paul RPGR.edi
Une proposition d’activité
- Argumenter, à partir de l’analyse de chacun de ces deus arbres, pour déterminer si le phénotype « rétinite » est récessif ou dominant et discuter de la localisation autosomale ou non du gène en cause.
- En confrontant les conclusions aux informations sur les gènes pouvant ête cause de rétinites, indiquer le ou les gènes qui pourraient être en jeu dans ces deux familles. Tester ces propositions l’aide des séquences relatives aux génotypes de quelques personnes des deux familles.
- Rechercher les conséquences des mutations identifiées dans ces deux familles.
- En cas d’union entre la femme III2 de la famille Pierre et l’homme III5 de la famille Paul, évaluer le risque de naissance d’un enfant qui au cours de sa vie sera victime d’une rétinite pigmentaire.
b - Les apports de l’analyse génotypique
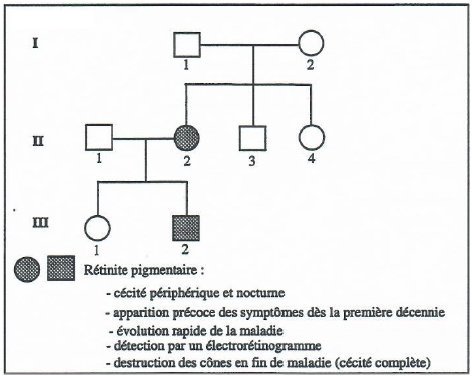
Arbre généalogique de la famille Michel
Cécité périphérique et nocturne apparaissant très précocement au cours de la vie, évolution rapide de la maladie
Généalogie famille Michel - Rhodpsine.edi
Généalogie famille Michel - Phosphodiestérase.edi
Arbre généalogique de la famille Martin
Généalogie famille Martin. Rhodopsine.edi
Généalogie famille Martin. Phosphodiestérase.edi
Une proposition d’activité
Argumenter, à partir de l’analyse de chacun de ces deus arbres, pour déterminer si le phénotype « rétinite » est récessif ou dominant et discuter de la localisation autosomale ou non du gène en cause.
En confrontant les conclusions aux informations sur les gènes pouvant être cause de rétinites, indiquer le ou les gènes qui pourraient être en jeu dans ces deux familles. Tester les propositions à l’aide des séquences relatives aux génotypes de quelques personnes des deux familles.
En conclusion, résumer comment l’analyse génotypique, orientée par l’analyse de l’arbre généalogique peut confirmer, préciser et même modifier les conclusions de cette dernière.
Indiquer la nature des mutations identifiées dans ces deux familles.
En cas d’union entre la femme II4 de la famille Michel et l’Homme III2 de la famille Martin, quelle est la probabilité que ce couple ait un enfant qui sera atteint de rétinite pigmentaire ?
III. Les raisonnements que les élèves doivent tenir
a - Hérédité autosomale - Hérédité liée au sexe
1. A propos de l’arbre généalogique de la famille Pierre.
- Le phénotype rétinite se retrouve à chaque génération ce qui suggère qu’il est dominant. Cela est confirmé par les phénotypes de I1, I2 et II2. L’homme II2 n’est pas malade alors que ses deux parents sont atteints de rétinite. Cela signifie que le phénotype « vision normale » est récessif et le phénotype « rétinite » dominant.
- Le gène n’est pas situé sur une région propre au chromosome Y car dans ce cas aucune femme ne devrait être atteinte de rétinite.
Si le gène est localisé sur une région propre au chromosome X, l’homme II2 a pour génotype X r+ Y et donc sa mère I1 et son père ont respectivement les génotypes XrXr+ (r : allèle à l’origine de la rétinite et r+, allèle non muté) et Xr Y. Les phénotypes de la troisième génération s’expliquent de même si II1 est hétérozygote et a le génotype XrXr+.
Les phénotypes de l’arbre généalogique s’interprètent donc très bien en admettant que le phénotype rétinite dans cette famille est dominant et que le gène en cause est localisé sur le chromosome X.
Mais une autre interprétation est possible : le gène en cause dans cette famille est situé sur un autosome. Dans ce cas, l’homme II2 a pour génotype r+//r+ et ses deux parents r//r+. le reste de l’arbre généalogique s’interprète très bien selon cette hypothèse (II1 et II3 hétérozygotes r//r+).
Deux interprétations sont donc possibles.
- Les renseignements sur les gènes impliqués dans les rétinites indiquent qu’on ne connait pas de rétinite liée au sexe dominante. Cela n’exclut pas obligatoirement cette possibilité si dans cette famille la rétinite est due à une mutation encore inconnue d’un gène situé sur le chromosome X.
Les données indiquent que des mutations de deux gènes peuvent causer des rétinites autosomales dominantes : celles affectant le gène Rho et celle du gène RDS. Ces deux gènes sont situés sur un autosome (chromosomes 3 et 6 respectivement). L’arbre généalogique ne permet pas de choisir.
- L’analyse moléculaire du gène RDS indique que toutes les personnes de la famille Pierre atteintes de rétinite ont un génotype avec deux allèles non mutés (donc de référence) de ce gène. Leur rétinite n’est donc pas due à des mutations de ce gène.
En revanche, pour le gène Rho, toutes les personnes atteintes de rétinite ont un génotype avec un allèle muté alors que les membres de la famille ayant une vision normale n’ont pas dans leur génotype d’allèle muté. Cela permet de conclure que dans cette famille Pierre, la rétinite est due à des mutations du gène Rho. En outre les personnes de génotype r+//r souffrent de rétinite ce qui confirme que le phénotype « rétinite » (et donc l’allèle r) est dominant dans cette famille.
- On constate aussi que deux mutations sont en jeu. La première présente chez I1, II1 et III2 au codon CGG135TGG, ce qui entraîne en position 135 de la rhodopsine la substitution de l’acide aminé arginine par la leucine (Arg135Leu). La deuxième présente chez I2, II3 et III3 consiste en une délétion de 4 codons (CTG68, CGC69, ACG70 et CCT71), ce qui entraîne dans la séquence de la rhodopsine la délétion de 4 acides aminés : Leu68, Arg69,Thr 70 et Pro71.
2. A propos de l’arbre généalogique de la famille Paul
- III3 et III5 sont atteints de rétinite alors que leurs parents ont une vision normale. Cela indique que dans cette famille, le phénotype rétinite est récessif.
- Le gène n’est pas situé sur le chromosome Y car dans ce cas le fils d’un homme atteint de rétinite devrait l’être aussi ce qui est infirmé par l’arbre (exemple : III7, IV3).
- Si le gène est localisé sur un locus propre au chromosome X, l’homme I1 a comme génotype Xr Y (r désignant un allèle du gène à l’origine de rétinite). Dans ce cas, toutes ses filles qui ont une vision normale ont comme génotype Xr Xr+. Les femmes II3, II5 et II7 ont donc ce génotype et elles ont pu transmettre à leurs fils III3, III5, III7, III10 et III12 leur chromosome Xr ce qui explique la rétinite de ces hommes.
Les phénotypes de cet arbre généalogique s’interprètent bien par une hérédité liée au sexe récessive.
- Si le gène est situé sur un autosome, le parent I1 a pour génotype r//r. En conséquence les femmes II3, II5 et II7 à vision normale ont pour génotype r+//r. Pour avoir des fils souffrant de rétinite, il faut que leurs conjoints, en dehors de la famille, soient aussi hétérozygotes de génotype r+//r. Etant donné la faible fréquence des allèles à l’origine de la maladie dans les populations, c’est peu probable mais pas impossible.
- L’analyse de l’arbre généalogique de la famille Paul indique donc que l’hypothèse la plus probable est que le gène en cause est situé sur le chromosome X.
Les informations fournies indiquent que le seul gène connu situé sur le chromosome X dont les mutations sont cause de rétinite est le gène RPGR.
L’analyse moléculaire du gène RPGR dans la famille indique que tous les hommes atteints de rétinite ont un génotype avec un seul allèle et que cet allèle est muté. Les hommes à vision normale ont uniquement l’allèle non muté du gène. Les femmes mères de garçons victimes de rétinite ont un chromosome X porteur d’un allèle normal et l’autre chromosome X avec l’allèle muté.
Les génotypes des membres de la famille confirment donc que la rétinite est due à des allèles mutés du gène RPGR.
La mutation présente dans cette famille est une mutation faux sens qui transforme le codon 514TCA codant pour la sérine en un codon stop TGA. La protéine est donc tronquée et non fonctionnelle.
- Risque que le couple a d’avoir un descendant atteint de rétinite
L’élève doit d’abord prendre en compte que les allèles de deux gènes sont en jeu, les allèles du gène Rho et ceux du gène RPGR. Il doit en conséquence indiquer le génotype des parents et préciser les symboles utilisés :
Pour le gène Rho, allèle RHO r+ et allèle Rho r
Pour le gène RPGR : allèle X r+ et Allèle X r
Pour les deux gènes, r désigne un allèle muté à l’origine de rétinite et r+ un allèle permettant une vision normale.
Le génotype de la femme III2 de la famille Pierre est donc :
Rho r//Rho r+, X r+// X r+
Le génotype de l’homme III5 de la famille Paul est donc :
Rho r+//Rho r+, X r Y.
Les deux gènes étant situés sur deux chromosomes différents, il doit appliquer ses connaissances sur le brassage interchromosomique pour déterminer les génotypes des gamètes produis par l’homme III5 et la femme III2. Il doit ensuite construire un échiquier de croisement prévisionnel.
L’homme produit deux types de gamètes : Rho r+/X r/ et Rho r+/Y/
La femme produit aussi deux types de gamètes Rho r/X r+ et Rho r+/ Xr+/
Il y a donc 4 génotypes possibles dans la descendance, deux pour les femmes et deux pour les hommes.
Du fait de la dominance de l’allèle muté du gène Rho et de la récessivité de l’allèle muté du gène RPGR, la probabilité de naissance d’un individu atteint de rétinite est de ½ tant en ce qui concerne les femmes que les hommes.
b. Analyse généalogique - analyse génotypique
1. A propos de la famille Michel
- II3 atteinte de rétinite a un phénotype différent de ses deux parents qui ont une vision normale. Dans cette famille, le phénotype « rétinite » est donc récessif.
En outre le gène n’est pas situé sur le chromosome Y car dans ce cas seuls les garçons peuvent avoir le phénotype « rétinite ».
Si le gène en cause est localisé dans la région propre au chromosome X, l’homme I1 dont la vision est normale possède un allèle non muté du gène situé sur le chromosome X. Dans ce cas, II3 qui a obligatoirement reçu le chromosome X de son père, ne peut avoir le phénotype rétinite qui est récessif. L’hypothèse est réfutée. Il ne reste plus que l’hypothèse d’une hérédité autosomale récessive. Dans ce cas, les deux parents I1 et I2 sont hétérozygotes. Ce qui explique les deux phénotypes trouvés dans la deuxième génération. - En confrontant cette conclusion avec les données sur les gènes dont les mutations engendrent le phénotype rétinite, il apparait que le gène en cause peut être soit le gène PDE, soit le gène Rho.
- L’analyse des séquences du gène PDE des membres de la famille indique qu’ils possèdent tous deux allèles non mutés. En conséquence, l’analyse moléculaire aboutit à la conclusion que ce gène n’est pas impliqué dans la rétinite de cette famille.
En revanche, pour le gène Rho, la femme II3 possède deux allèles mutés, et I1, I2 et II4 un allèle normal et un allèle muté.
Les techniques de séquençage permettant de connaître le génotype des personnes a donc permis, par rapport à l’analyse généalogique, de préciser le gène cause de rétinite dans cette famille, le gène codant pour la rhodopsine.
La mutation à l’origine de l’allèle muté est une substitution non-sens au codon 249 : GAG249TAG. Le codon TAG est un codon stop. La rhodopsine résultant de l’expression de cet allèle muté est donc raccourcie (248 acides aminés au lieu de 348) et non fonctionnelle.
2. A propos de la famille Martin
- La femme II2 est atteinte de rétinite alors que ses parents ont une vision normale. Dans cette famille, le phénotype « rétinite » apparait donc récessif. Toutefois, le fait que le fils III2 de cette femme ait aussi une vision déficiente engendre un doute sur cette récessivité.
- Le gène en cause ne peut être situé sur la région propre au chromosome Y car dans ce cas une femme ne peut être atteinte de rétinite. Si le gène est localisé sur le chromosome X II2 a le génotype Xr Xr. Dans ce cas, le père de II2, I1 ne peut avoir une vision normale car il possèderait l’allèle r. Cette hypothèse est réfutée.
Il reste la possibilité que le gène soit localisé sur un autosome. L’arbre est en accord avec cet hypothèse à condition que II1 soit hétérozygote et possède donc un allèle muté du gène en jeu.
- En confrontant ces conclusions avec les informations sur les gènes dont les mutations causent des rétinites, on arrive à deux possibilités : dans cette famille, la rétinite est due à une mutation du gène PDE ou du gène Rho (avec un allèle cause d’une rétinite récessive).
- L’analyse des génotypes montre que tous les individus dont le génome a été analysé, en particulier II2, possèdent deux allèles « normaux » du gène PDE. La rétinite de cette famille n’est pas due à ce gène.
II2 possède un allèle muté et un allèle normal du gène Rho. Cela conforte l’idée que le gène Rho est en jeu dans cette famille mais réfute l’idée de rétinite récessive.
I1 et I2 ont un génotype avec deux allèles normaux du gène Rho. Cela réfute l’idée qu’ils étaient hétérozygotes pour le gène Rho.
Pour expliquer que I1 et I2 aient un génotype avec deux allèles normaux du gène Rho alors que leur fille II2 possède un allèle muté, il faut admettre une néomutation (mutation de novo) lors de la formation des gamètes chez un des parents.
Cela est confirmé par le fait que III2 possède le même allèle muté que sa mère II2. - La détermination des génotypes par les techniques de séquençage a donc nettement modifié l’interprétation résultant de l’analyse de l’arbre généalogique : la rétinite dans cette famille est dominante et non récessive et son apparition chez des membres de la famille est due à une néomutation.
La néomutation est une substitution faux sens qui transforme le codon CCG en position 347 en un codon CTG (CCG347CTG) ce qui, au niveau de la rhodopsine, se traduit par la substitution de l’acide aminé leucine à l’acide aminé proline (Pro347Leu).
Risque qu’a le couple II4 (famille Michel) - III2 (famille Martin) d’avoir un descendant ayant une rétinite.
Dans les deux familles, c’est le même gène, le gène RHO, qui est en cause mais les mutations sont différentes entre les deux familles.
Il faut indiquer les génotypes de II4 et III2 en précisant les symboles utilisés.
Génotype II4 : Rho249//Rho r+. (Rho r+ indique un allèle fonctionnel du gène, Rho249 désigne un allèle muté et la position du codon muté dans la séquence).
Génotype de III2 : Rho r+// Rho347
Il suffit de construire l’échiquier de croisement prévisionnel de la descendance du couple et de prendre en compte dominance et récessivité des allèles pour arriver à la conclusion qu’il y a une probabilité de ½ de descendant atteint de rétinite.