
Copie-Origine et évolution du génome de Sars-CoV-2
1 . Vue globale des génomes des coronavirus humains
Le Sars-CoV-2 (Sars-CoV pour : Severe acute respiratory syndrome coronavirus) est le septième coronavirus humain connu. Tous les 7 causent des maladies respiratoires. Quatre d’entre eux OC43 (HCoV-OC43), 229E (HCoV-229E), NL63 (HCoV-NL63) et HKU1 (HCoV-HKU1) causent des maladies relativement bénignes, s’apparentant à un rhume plus ou moins fort. En revanche les trois autres peuvent être à l’origine de syndromes respiratoires aigus, sévères pouvant être mortels
Le Sars-CoV-1 est un virus émergent apparu en Chine en 2002 et ayant causé de nombreux cas de pneumonie grave jusqu’au milieu de l’année 2003. Le Mers-CoV (Mers pour : Middle East Respiratory Syndrome) est apparu au Moyen-Orient en 2012 (premier cas en Arabie saoudite) et resté confiné aux milieux hospitaliers car il ne semble pas se propager aisément d’une personne à l’autre. Enfin, le Sars-CoV-2 détecté en premier à Wuhan en chine, en décembre 2019, se transmet facilement d’un humain à un autre et est à l’origine de la pandémie COVID-19.
Ainsi, en une vingtaine d’années, trois nouveaux coronavirus capables de parasiter l’organisme humain et pouvant causer des pneumonies graves sont apparus. Rechercher l’origine de ces virus est un objectif dans la mesure où cela peut déboucher sur les attitudes à suivre pour en éviter l’apparition de nouveaux.
Le Sars-CoV-2 parmi les coronavirus humain
Le Sars-CoV-2 est le dernier coronavirus humain apparu. On peut imaginer d'abord qu’il provient de l’évolution d’un coronavirus humain préexistant, évolution le rendant plus pathogène.
Pour tester cette hypothèse, on dispose des génomes des 7 coronavirus humains.
Fichier pour Anagène : Coronavirus humains-Hcov-bis.edi
Fichier pour Phylogène : Coronavirus-humains-Hcov-bis.aln
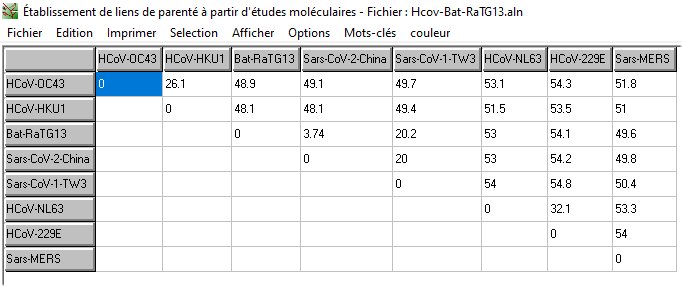
La comparaison deux à deux des séquences du génome de ces virus avec Anagène, permet d’établir une matrice des distances révélant la similitude globale plus ou moins grande de leurs génomes. A partir de cette matrice, on peut construire un arbre phylogénétique reposant sur l’idée que plus les séquences des génomes de deux virus sont similaires, plus ils sont étroitement apparentés.
On peut également obtenir cette matrice et l'arbre phylogénétique correspondant avec Phylogène.
Hcov-Arbres et matrice des distances
On constate que c’est avec la Sars-CoV-1 que le Sars-CoV-2 est le plus étroitement apparenté et non avec un coronavirus « bénin ». Cela est déjà un argument fort contre l’idée que le Sars-CoV-2 provient de l’évolution d’un coronavirus humain préexistant.
Question : peut-il provenir de l'évolution de Sars-CoV-1 ?
La similitude des Sars-CoV-1 et Sars-CoV-2 est de l’ordre de 80%. Les génomes de ces coronavirus ayant une longueur de 30.000 nucléotides environ, cela signifie qu’ils diffèrent par près de 6000 nucléotides. Comme le taux des mutations du génome de ces coronavirus est estimé à 25 mutations pour l’ensemble du génome et par an, cela exclut que le Sars-CoV 2 puisse provenir [directement] d’une évolution du Sars-CoV1. Il faut donc rechercher une autre origine au Sars CoV-2.
Comme les virus sont obligatoirement des parasites, on est conduit à l’idée que c’est un coronavirus parasitant une espèce animale qui est à l’origine du Sars-CoV-2 (comme de tous les autres coronavirus...).
2 . Les Chauve-souris, des réservoirs à virus
Plusieurs espèces peuvent être parasitées par des coronavirus mais ce sont les chauve-souris qui sont de très loin les plus infectées, ce qui fait qu’on les considère comme des réservoirs à virus. Chose remarquable, les chauve-souris ne manifestent généralement pas de signes cliniques associés avec l’infection.
Il existe de très nombreux coronavirus de Chauve-souris, chacun étant plus ou moins spécifique d’une espèce. Les génomes de ces coronavirus ont été séquencés. C’est ainsi qu’en 2013, on a séquencé celui du coronavirus RaTG13 qui parasite l’espèce de chauve-souris Rinolophus affinis.
On dispose de la séquence de ce génome ce qui permet de le comparer au génome du Sars-CoV-2. On trouve une identité entre les deux génomes ce qui permet de compléter l’arbre phylogénétique précédent en intégrant le génome de RaTG13.
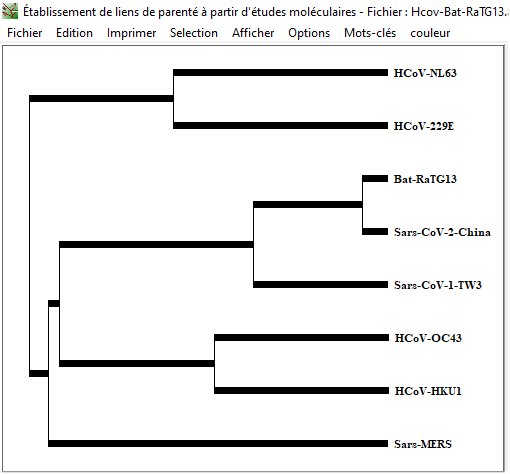
Génomes de coronavirus de chauve-souris (un extrait d’arbre de Nextstrain.)
Les séquences de RaTG13 et de Sars-CoV-2 ont une identité beaucoup plus forte que celle des autres coronavirus humains avec Sars-CoV-2. Cela laisse supposer que le RaTG13 parasitant Rinolophus affinis ou du moins certains représentants de ce virus pourraient avoir subi une évolution leur permettant un saut d’espèce, c’est-à-dire la possibilité d’infecter des cellules humaines.
Pour tester l’idée qu’un coronavirus animal peut être à l’origine d’un coronavirus humain, on peut faire le point des connaissances sur l’origine du Sars-CoV-1, agent de l’épidémie de 2002-2003 .
3 . L’origine du Sars-CoV-1
Les études épidémiologiques lors de l’épidémie du Sars-CoV-1 en Chine ont attiré l’attention sur le fait que beaucoup de personnes infectées avaient visité des marchés où on vendait des animaux vivants. Parmi ceux-ci des Civettes palmées. Ces civettes sont des carnivores sauvages qui ont commencé à être domestiqués pour leur fourrure depuis la fin des années 50, puis élevés pour servir de nourriture exotique dans des restaurants. En 2003, on estimait que 600 fermes en Chine faisaient l’élevage de 40.000 civettes en tout.
Les chercheurs ont supposé que plusieurs personnes infectées par le Sars-CoV-2 l’avaient été par l’intermédiaire d’un coronavirus transmis par des Civettes. Autrement dit, un coronavirus présent chez les Civettes devait avoir franchi la barrière d’espèce et devenu capable de parasiter les cellules de l’appareil respiratoire humain. Ils ont alors identifié les coronavirus présents chez des Civettes et séquencé leurs génomes.
Cependant, ils ont constaté que de nombreuses Civettes n’hébergeaient pas de coronavirus, ce qui laissait penser que les coronavirus hébergés par les Civettes infectées provenaient d’autres espèces animales. La recherche s’est orientée vers les chauve-souris, connues pour être des réservoirs à virus. Ils ont trouvé que plusieurs espèces du genre Rhinolophus étaient parasitées par des coronavirus dont ils ont séquencé le génome.
Les documents indiqués ci-dessous permettent de proposer le sujet de recherche suivant :
En quoi les informations extraites de l’exploitation des données génomiques corroborent l’affirmation suivante : « Certaines espèces de Chauve-souris hébergent des coronavirus qui ont pu être à l’origine du Sars-CoV-1 mais la transmission du virus de la chauve-souris n’a pas été directe mais a impliqué au moins un hôte intermédiaire, la Civette palmée.
Documents
Séquence nucléique du Sars-CoV-1 et Séquence nucléique du coronavirus de la civette (civet007)
Fichier Sars-CoV-1-Tw3-Sars-Civet007.edi
Ces chercheurs ont comparé les séquences de 10 génomes de coronavirus trouvés chez des Civettes. Ils ont repéré 26 sites pour lesquels les 10 génomes avaient le même nucléotide. Ils ont analysé les génomes du Sars-CoV-2 humain prélevés au début de l’infection jusqu’à la fin. Ils ont identifié le nucléotide présents à chacun des 26 sites.
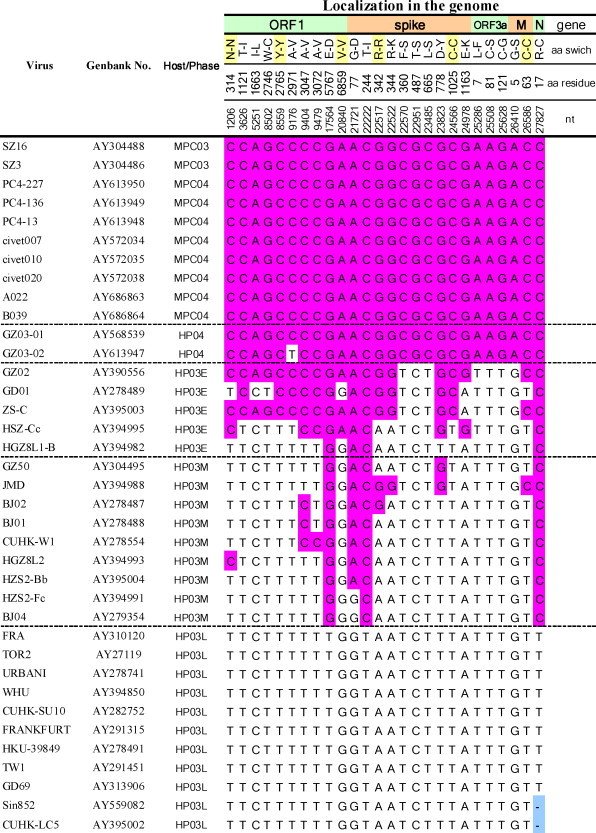
Table 2 de l'article : A review of studies on animal reservoirs of the Sars coronavirus. Virus research 2008 Zhengli Shi ; Zhihong Hu.
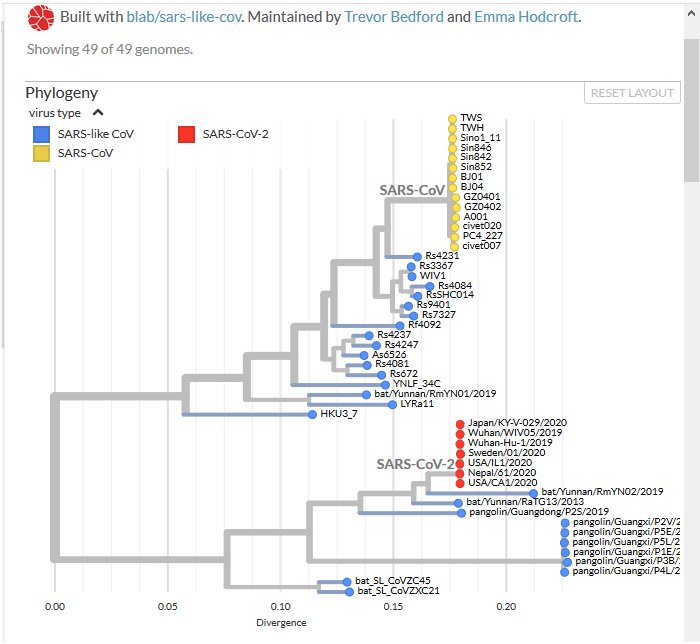
Arbre phylogénétique général des coronavirus (Phylogeny of Sars-like betacoronavirus including novel coronavirus Sars-CoV-2. Nextstrain.)
Les génomes des Sars-CoV-1 et des coronavirus de civettes sont en jaune, ceux trouvés chez des chauve-souris rhinolophes sont en bleu et ceux du Sars-CoV-2 en rouge.
Génomes de coronavirus trouvés chez des Chauve-souris Rhinolophes : WiV1 ; Rs3367 ; RsSHC014
Exploitation de ces documents
La comparaison avec Anagène des génomes de référence du Sars-CoV-1 humain et du coronavirus de la civette (civet007) indique que la similitude de ces deux génomes est supérieure à 99, 6% ce qui montre une très étroite parenté. L’arbre phylogénétique des coronavirus confirme que cela est vrai pour tous les génomes Sars-CoV1 et de Civette séquencés. Cela indique que les Sars-CoV-1 à l’origine de l’épidémie de 2002-2003 ont pour origine des coronavirus parasites de civettes comme le laissaient penser les études épidémiologiques.
La table 2 de l'article : A review of studies on animal reservoirs of the Sars coronavirus. Virus research 2008 Zhengli Shi ; Zhihong Hu montre une évolution du génome du Sars-CoV-1 au cours de l’infection. Au début de l’infection pour les 26 sites considérés, les génomes des Sars-CoV-1 sont identiques à ceux des Civettes. Ensuite, au fur et à mesure que progresse l’infection, les séquences diffèrent de plus en plus de celles des coronavirus des Civettes et, en fin d’infection, ils diffèrent à tous ces sites. Cela traduit une évolution du génome du Sars-CoV-1 chez l’Homme.
Les premiers Sars-CoV étaient des coronavirus transmis par les Civettes. Ensuite s’est installée une transmission interhumaine. Des mutations au cours de la propagation de la zoonose chez l’homme ont fait évoluer les génomes du Sars-CoV-1.
[Ces mutations sont des substitutions et certaines affectent le génome de la protéine « spike » cruciale pour l’entrée du génome du virus dans les cellules humaines. On a montré que la mutation 22951 du génome entraînait en position 487 de la protéine S, la substitution de l’acide aminé thréonine à l’acide aminé sérine. Or à cette position est importante pour la liaison entre la protéine S et le récepteur cellulaire ACE2 des cellules humaines qui conditionne l’entrée du virus dans les cellules humaines. On a montré que cette substitution facilite l’entrée du virus dans les cellules humaines. Cette évolution du Sars-CoV-1 chez l’homme est la traduction de l’intervention de la sélection naturelle dans cette évolution.]
La comparaison avec Anagène des génomes des Sars-CoV humains et de Civettes avec ceux des coronavirus, RsSHC014, Rs3367, WIV1, de chauve-souris rhinolophes de la grotte de Yunan en Chine montre une similitude globale de 90% environ. Cette similitude est nettement supérieure à celle trouvée pour d’autres coronavirus de Chauve-souris. En outre ces virus utilisent aussi le récepteur ACE2 des cellules pour les parasiter. Cette similitude est bien illustrée par l’arbre phylogénétique qui montre ces coronavirus de Chauve-souris étroitement apparentés aux Sars-CoV humains et de Civettes.
L’ensemble de ces données confirme que le Sars-CoV-1 a une origine animale. Sa source semble être certains coronavirus infectant des chauve-souris rhinolophes sans provoquer de maladies chez ces animaux. L’évolution de leur génome chez les chauve-souris leur a conféré la possibilité d’infecter une espèce de mammifères, les Civettes. Celles-ci ont joué le rôle d’hôte intermédiaire entre les chauve-souris et l’homme. Le Sars-CoV-1 transmis par les Civettes a continué à évoluer chez l’homme (mutations) et la sélection naturelle a conduit à des populations de virus se multipliant efficacement chez les humains infectés.
4 : L’origine du Sars-CoV-2 responsable de la pandémie Covid-19
Pour aborder ce sujet, on dispose des documents suivants : A faire. En cours
- Séquences des génomes (complets) du Sars-CoV-2, du coronavirus RaTG13 infectant une Chauve-souris rhinolophe, des coronavirus de Chauve-souris : bat_SL_CoV ZC45, et bat_SL_CoV ZXC21, de Pangolin Guangdong.
Fichier Sars-divers bat et pangolin.edi
- Phylogeny of Sars-like betacoronavirus including novel coronavirus Sars-CoV-2 Nextstrain
Cet arbre phylogénétique global est déjà dans le paragraphe précédent.
- Séquences nucléiques du gène S de Sars-CoV-2, RaTG13 et du Pangolin. Séquences protéiques S des mêmes coronavirus.
Fichier Spike-Sars-CoV2-Sars-Pangolin-Sars-RaTG13-ADN-Pro.edi
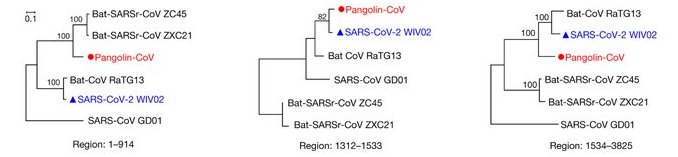
- Phylogénies (arbre) de Sars-CoV2, RaTG13 et Pangolin CoV construites à partir de données sur différentes régions du gène S ;
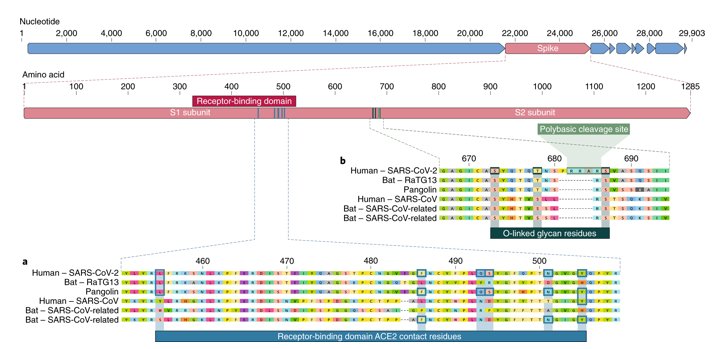
- Séquences du RBD des protéines S du Sars-cov-2, de RaTG13 avec mention des acides aminés cruciaux pour la liaison au récepteur cellulaire ACE2.
Exploitation pédagogique
-
La recherche sur l’origine du Sars-CoV-1 a abouti à l’idée que c’est un coronavirus hébergé par une espèce de Chauve-souris qui était à l’origine du coronavirus humain responsable de l’épidémie 2002-2003. Toutefois la transmission du coronavirus de la Chauve-Souris à l’Homme n’a pas été directe mais a impliqué au moins une espèce intermédiaire la Civette. Dès l’élucidation du génome du Sars-CoV-2 en janvier 2020, des chercheurs se sont attaqués au problème de l’origine de ce virus humain émergent et, se basant sur les connaissances acquises à propos du Sars-CoV-1, ils ont cherché des coronavirus de Chauve-souris qui pourraient être à l’origine du nouveau virus humain. Ils ont ainsi séquencé le génome de 3 coronavirus : RaTG13, SlZc45 et SLZXC21 présents chez des Chauve-souris. D’autres chercheurs dont l’attention a été attirée par des informations suivant lesquelles des pangolins manifestaient des symptômes proches de ceux du Covid-19 humain, ont trouvé des coronavirus chez ces animaux malades et séquencé les génomes de ces virus.
-
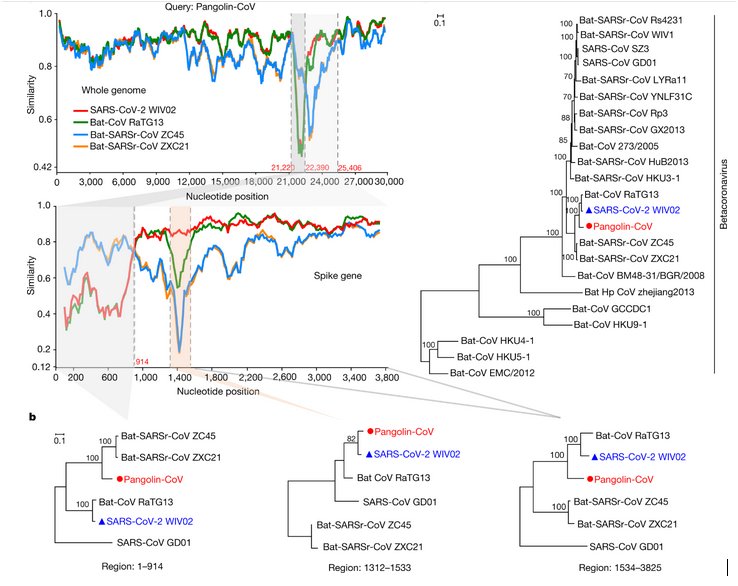
La comparaison avec Anagène des séquences des génomes des 3 coronavirus de chauve-souris et de celui trouvé chez les pangolins avec le génome de Sars-CoV-2 permet donc de discuter si ces coronavirus animaux peuvent être à l’origine du virus humain. On constate que c’est le génome du RaTG13 qui a la plus forte similitude – (plus de 96% d’identité) avec le Sars-CoV-2 ce qui est un fort argument pour dire qu’il peut être à l’origine du Sars-CoV-2. Le virus du Pangolin a aussi une forte similitude avec le virus humain de l’ordre de 90%. Les coronavirus ZC45 et ZXC21 de Chauve-souris ont une plus faible identité avec le Sars-CoV-2.
Cette comparaison peut se prolonger par la réalisation d’un arbre phylogénétique simple relatif à ces virus qu’on peut confronter avec l’arbre beaucoup plus complet fourni par le site « Nextstrain ». (Phylogeny of sars-like betacoronavirus including novel coronavirus Sars-CoV-2.) -
Une analyse plus fine du gène qui code pour la protéine S qui permet la fixation du coronavirus à des récepteurs cellulaires (ACE2 pour le sars-CoV-2 ) des cellules de l’animal infecté, fournit des informations sur une modalité de l’évolution des génomes des coronavirus. Pour cela il faut comparer avec Anagène les séquences nucléotidiques des gènes S dans trois régions différentes : de 1 à 914 nucléotides, de 1312 à 1533 nucléotides et de 1534 à 3825 nucléotides.
-
On constate que le génome du pangolin est plus similaire à ceux des coronavirus ZXC21 ZC45 qu’à ceux du RaTG13 et du Sars-CoV-2 dans la première partie (1-914) du gène. En revanche dans la région 1312-1533 la séquence du coronavirus du Pangolin est plus proche de celle du Sars-CoV-2 que des coronavirus de Chauve-souris, RaTG13 compris. Enfin dans la dernière partie 1533- 3825, on retrouve la même phylogénie que celle fournie par la comparaison des génomes globaux. Cela est traduit par les 3 arbres phylogénétiques ci-dessous.
-
Ainsi le gène S du coronavirus du Pangolin, possède une région qui semble provenir du coronavirus ZC45 ou ZXC21 et une région ayant pour origine le RaTG13. Cela traduit une recombinaison génétique associant des fragments de deux génomes différents. Cela est possible si deux virus différents infectent les mêmes cellules et si cela est suivi d’un phénomène ayant lieu lors de la réplication des génomes viraux dans la cellule parasitée.
Ainsi l’évolution du génome des coronavirus peut se faire par mutation et par recombinaison génétique ; cette dernière modalité a des effets plus drastiques que la mutation. -
La comparaison du gène de la protéine S indique que dans la région 1312-1533, la séquence du Pangolin a une identité nettement plus forte avec celle du Sars-CoV-2 que ne l’a la séquence du RaTG13. Or cette région correspond au RBD, c’est-à-dire à une région du gène S qui code pour le domaine de fixation au récepteur ACE2 des cellules humaines parasitées. Cela indique que le coronavirus RaTG13 malgré la très grande identité globale de son génome avec celui de Sars-CoV-2 ne peut être le virus qui, directement transmis à l’homme, a été à l’origine de Sars-CoV-2. Cela est confirmé par une analyse fine de ce domaine RBD. On a identifié 6 acides aminés du RBD de la protéine S du Sars-CoV-2 qui jouent un rôle crucial dans la liaison au récepteur ACE2 : L455, F486, Q493, S494, N501 et Y 505.
Le document ci-dessous renseigne sur les séquences des RBD des coronavirus précédemment envisagés.

-
On peut constater que le RBD de RaTG13 n’a qu’ un seul des 6 acides aminés contrairement au coronavirus du pangolin qui en possède les 6.
Cela indique que le coronavirus RaTG13 de la Chauve-souris Rhinolophus affinis ne peut être le virus qui a réussi à franchir la barrière d’espèce et causé la pandémie du Covid-19
Conclusion
-
Le coronavirus RaTG13 de la chauve-souris Rhinolophus affinis a un génome qui présente une identité avec celui du génome de Sars-CoV-2 de l’ordre de 96%, ce qui est supérieure à l’identité avec tous les coronavirus animaux et humains qui ont été séquencés. Cela traduit une étroite parenté entre RaRG13 et Sars-CoV-2 et laisse à penser que le Sars-CoV- 2 apparu en 2019 pourrait provenir du RaTG13 directement transmis à l’homme où il aurait évolué pour devenir la cause du Covid-19.
-
Une analyse précise des séquences du RaTG13 et du Sars-CoV-2 amène à mettre en cause cette filiation directe entre les deux coronavirus. En effet les deux génomes divergent dans une région précise de leur génome, celle qui code pour la protéine S ; Les protéines S du RaTG13 et du Sars-CoV-2 diffèrent en particulier dans leur domaine de liaison (RBD) aux récepteurs ACE2 des cellules humaines. Le RaTG13 ne peut se fixer aux récepteurs ce qui souligne qu’il ne peut être directement à l’origine du Sars-CoV-2.
-
Le coronavirus du Pangolin a une identité globale avec Sars-CoV-2 légèrement inférieure à 90% ce qui indique une parenté moins étroite avec Sars-CoV-2 que celle de RaTG13. Cependant, la région du génome du Pangolin qui code pour la protéine S et plus précisément son RBD a une très forte identité, supérieure à 97%, avec la région homologue du Sars-CoV-2. On a donc une situation opposée à celle trouvée avec le RaTG13. La séquence globale du génome du virus du pangolin est trop différente de celle du Sars-CoV-2 pour qu’il puisse être considéré comme l’ancêtre immédiat du Sars-CoV-2 comme le RBD du RaTG13 indique que ce virus ne l’est pas non plus.
-
Le Sars-CoV-2 doit être issu d’un coronavirus animal présentant un génome global identique ou très proche de celui du RaTG13 mais avec une région codant pour le RBD de la protéine S très proche de celle de la région homologue du coronavirus du Pangolin. Un tel virus ne peut que résulter d’une recombinaison génétique entre deux coronavirus. On ne sait si elle a eu lieu chez une chauve-souris ou chez un pangolin avant que le virus soit transmis à l’homme. Ensuite, le virus a évolué chez l’homme par des mutations et sous l’action de la sélection naturelle, est devenu de plus en plus capable de se multiplier efficacement
-
Les chercheurs chinois ont trouvé que dans des grottes du sud de la Chine vivaient des populations diverses de Chauve-souris, aux génomes différents. En outre, fréquemment les chauves-souris hébergent plus d’une sorte de coronavirus. Cela crée des conditions favorables à la recombinaison génétique et donc à l’émergence de nouveaux coronavirus. Mais les Chauve-souris ne transmettent généralement pas leurs coronavirus à l’homme mais à un hôte intermédiaire qui le transmet ensuite à l’Homme. Le risque est important lorsque l’hôte intermédiaire est un animal sauvage domestiqué par l’Homme. C’est le cas de la Civette pour l’épidémie du Sars-CoV1 de 2002. En revanche, pour le Sars-coV-2, on ne connait pas en 2020 quel est l’hôte intermédiaire.