
DM1 - Texte initial
Ancien texte
La maladie de Steinert
La maladie de Steinert encore appelée dystrophie myotonique (en abrégé DM1) est une maladie génétique héréditaire dont la prévalence est estimée à 1 pour 8000 personnes. Elle touche essentiellement les muscles squelettiques (affaiblissement et atrophie des muscles appelée dystrophie, difficulté au relâchement après la contraction appelée myotonie. Elle peut affecter d’autres organes, en particulier le cœur.
On distingue quatre formes de la maladie en fonction de la date d’apparition des premiers symptômes :
- la forme congénitale, à début néonatal ;
- la forme infantile ou juvénile ;
- la forme commune de l’adulte ;
- la forme à début tardif.
Les formes les plus précoces sont généralement les plus graves.
Le gène dont les mutations sont causes de cette maladie génétique, découvert en 1992, est le gène DMPK (pour dystrophine myotonine protéine kinase).
Il faudrait mettre là la problématique globale.
1 - Les caractéristiques de la transmission héréditaire du phénotype DM1
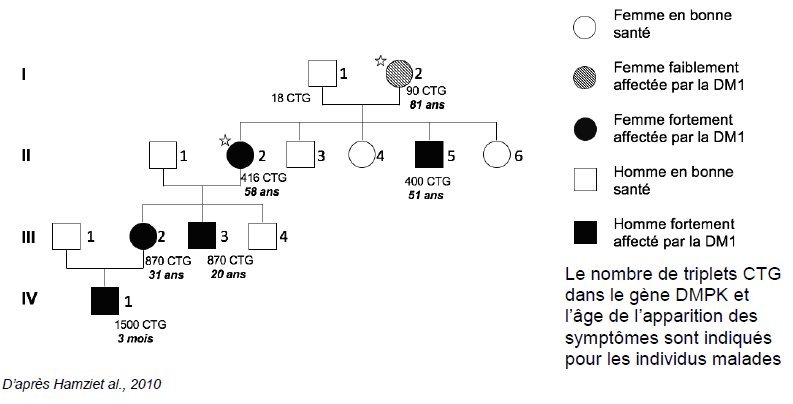
La figure 1 est celle de l’arbre généalogique d’une famille atteinte de la maladie de Steinert avec l’âge d’apparition des premiers symptômes.
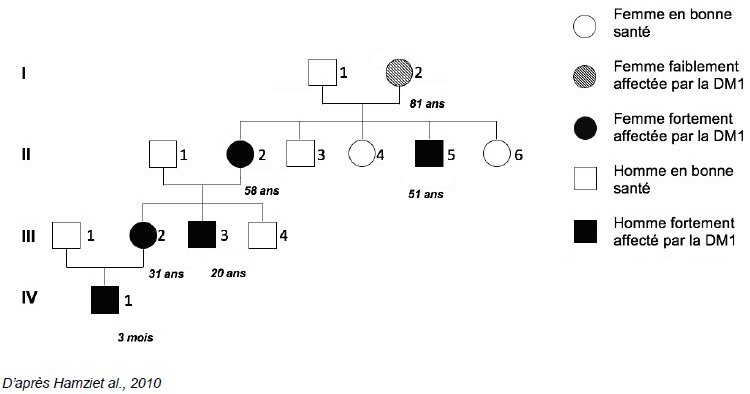
La figure est extraite de : Antropo 2010 23, 73-76 Hamzi et al. La maladie de Steinert dans une famille marocaine: phénomène d’anticipation et conseil génétique.
On peut poser les questions suivantes :
A partir de raisonnements reposant sur les informations fournies par l’arbre, indiquer :
- si le phénotype DM1 est récessif ou dominant ;
- si le gène DMPK est situé sur un autosome ou un chromosome sexuel ;
- le génotype des individus atteints par la maladie en désignant par m tout allèle cause de la maladie et par m+ tout allèle n’entraînant pas la DM1 ;
- préciser les caractéristiques de l’évolution de la maladie au fil des générations ;
- la figure suivante montre un arbre généalogique d’une famille atteinte par une myopathie.
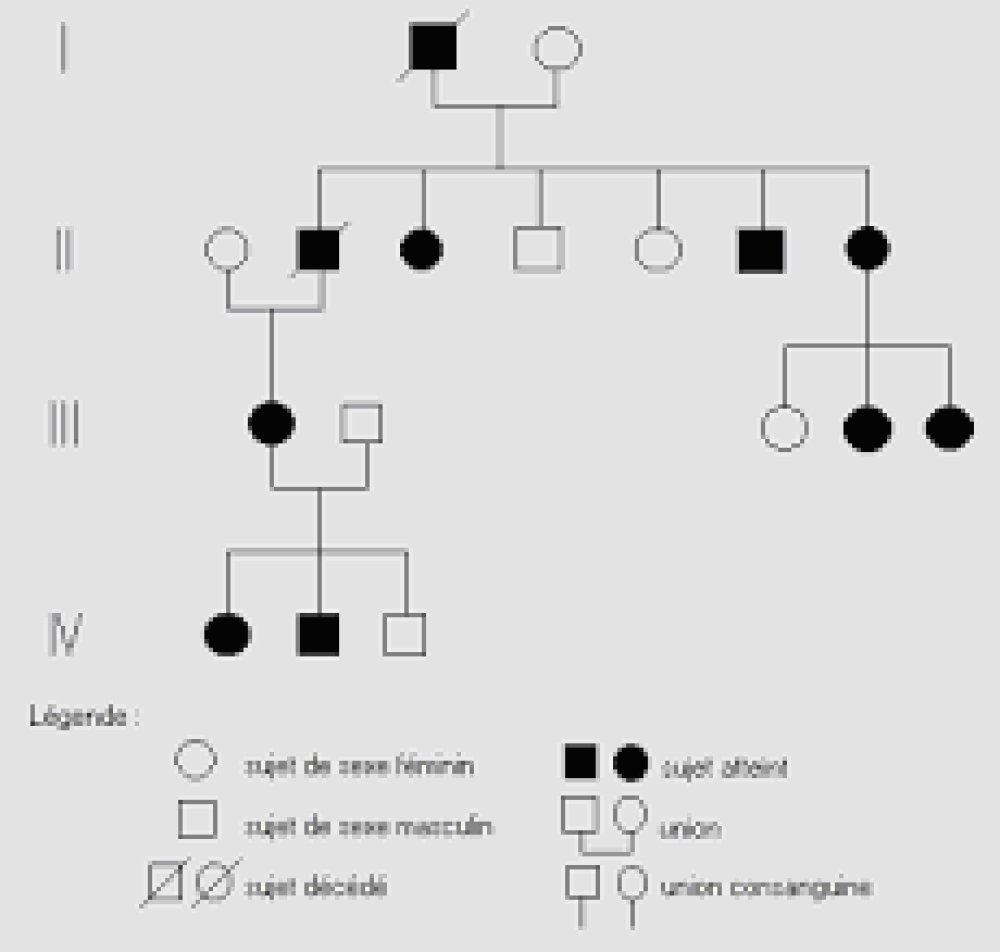
A partir des informations tirées de cet arbre généalogique, indiquer si cette maladie génétique est due à un mécanisme de transmission génétique semblable à la myopathie de Duchenne ou à la maladie de Steinert (Dominance ou récessivité, localisation chromosomique du gène en cause).
2 - Les caractéristiques génétiques du gène DMPK
A partir des données relatives aux séquences se rapportant au gène DMPK, il s’agit d’aboutir à une représentation schématique du gène DMPK ciblée sur les caractéristiques de ce gène ou plus exactement d’un allèle fonctionnel. Pour faciliter l’exploitation de ces séquences, un document de référence relatif au gène qui code pour la chaîne bêta de l’hémoglobine est fourni :
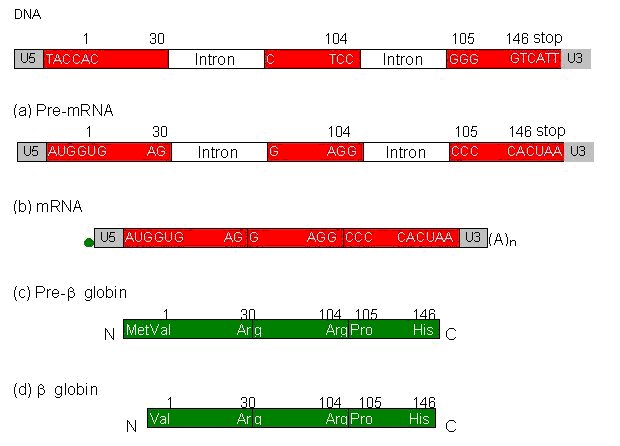
Cf : Génétique et évolution en première spécialité Expression de l’information génétique programme 2011. Notion de gène morcelé et d’épissage.
Rappel sur la synthèse des protéines donc sur l’expression des gènes. (AFMTELETHON juin 2022)
 |
Un ARN messager est une copie d'un gène. Il comporte l'information nécessaire pour fabriquer une protéine. 1. L'ARN pré-messager est produit dans le noyau à partir de la séquence d'ADN (transcription). 2. Il subit un processus de maturation (épissage) pour donner un ARN messager utilisable pour la synthèse d'une protéine. 3. L'ARN messager sort du noyau vers le cytoplasme où a lieu la synthèse des protéines. 4. Il est utilisé comme "patron" par la machinerie cellulaire pour fabriquer une protéine (traduction). Chacune de ces étapes est contrôlée par des protéines dites "régulatrices". |
L’enzyme ARN polymérase II assure la transcription du gène bêta au-delà du codon stop. Elle cesse cette transcription lorsqu’elle rencontre dans la séquence du gène, au-delà du codon stop, un signal de terminaison. Ce signal est la séquence AATAAA. La transcription est interrompue 10 à 30 nucléotides en aval de ce signal.
Après, au cours de cette maturation dans le noyau de l’ARN pré-messager, les introns sont éliminés et les exons liés les uns à la suite des autres. En outre une queue de nucléotides poly A est ajoutée à la fin de la séquence de l’ARN messager.
Les élèves pourraient aboutir à la représentation suivante de la structure du gène DMPK :
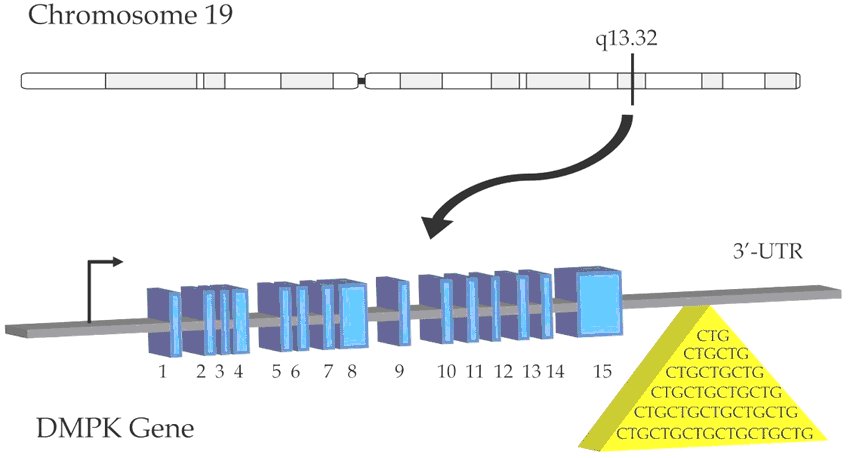
Source : Myotonic Dystrophy Type 1...
Les rectangles en bleu représentent les exons. Le schéma indique 15 exons. Cela doit résulter de la comparaison des séquences du gène et de l’ARN pré-messager produit immédiat de la transcription du gène.
Il faudrait situer le codon stop sur le 15ème exon. Surtout l’originalité de la structure de ce gène est visualisée en jaune : c’est une répétition du triplet CTG dans la séquence du gène qui est transcrite mais non traduite. Le nombre de répétitions d’une vingtaine est conforme à une expression normale du gène. On peut ajouter la fin de la transcription et situer le séquence AATAAA signal annonçant la fin de la transcription.
Les séquences de test
(Fichier DMPK.edi pour Anagène ou Géniegen 2).
Pour le moment tout est en ADN.
J'ai retenu le gène DMPK complet (19840 pb), le variant2 transcrit (Pré-messager, 2799 pb) et le CDS correspondant (1890 pb). Tels qu'ils figurent dans Genbank.
Voici quelques nouvelles représentations :
- Alignement du gène, du pré-messager et du CDS montrant la fin du 15ème exon

- Alignement montrant les répétitions de CTG dans le gène et le pré-messager

- Alignement montrant la position du AATAA dans le gène et dans le pré-messager

Noter : La mise en évidence de la séquence AATAAA dans le Pré-messager, la position du stop dans le 15ème codon et la présence de répétitions dans le pré-messager peuvent être localisées avec Anagène, pas avec Géniegen.
Pour la structure du gène :
Avec Géniegen2, on compare les deux séquences, on fait un dotplot et on compte les segments identiques. Résultat probant.
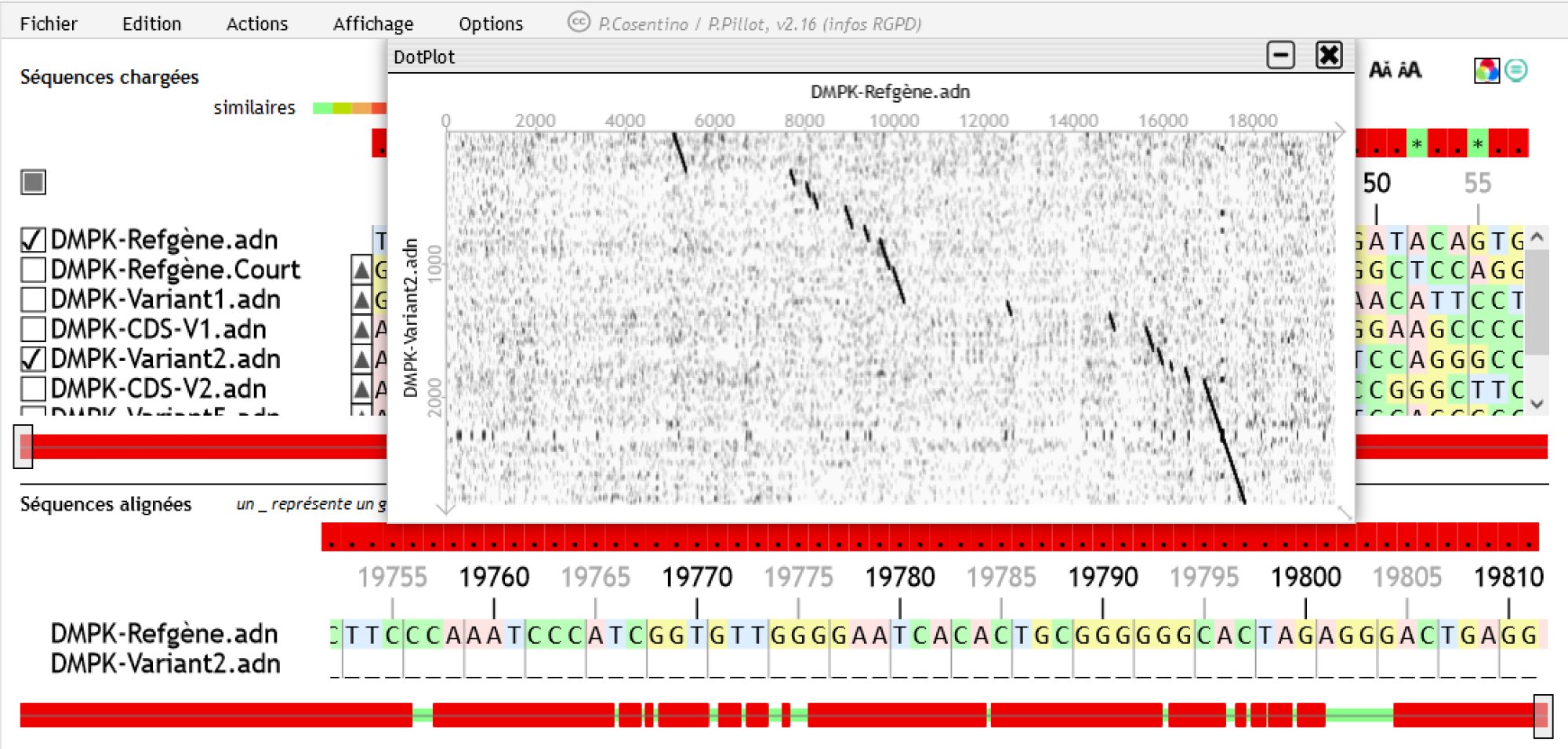
Avec Anagène on aligne les deux séquences et il faut parcourir le résultat et comper les segments identiques... C'est manuel et peut-être plus fastidieux mais instructif.
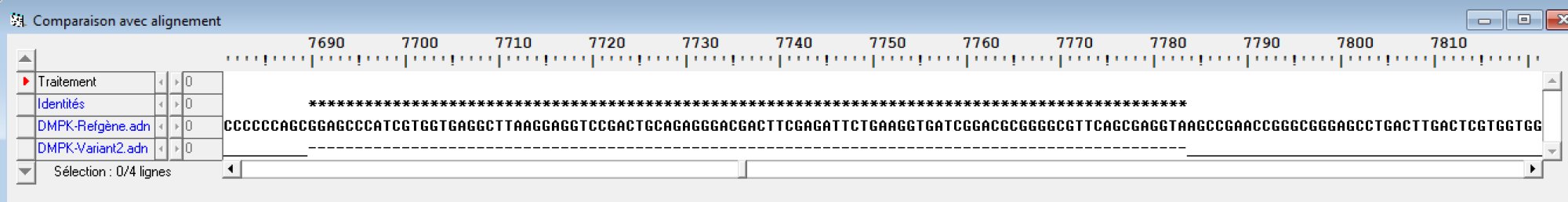
Position de AATAAA avec Anagène
On se place (on clique) au début de la séquence. Ensuite, Fichier, Rechercher. On rentre AATAAA. Résultat :
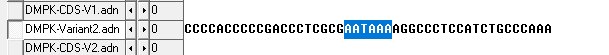
Position du stop avec Anagène
Comparaison du pré-messager et du CDS
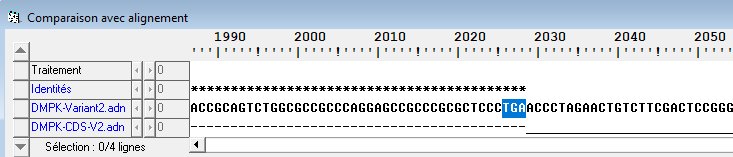
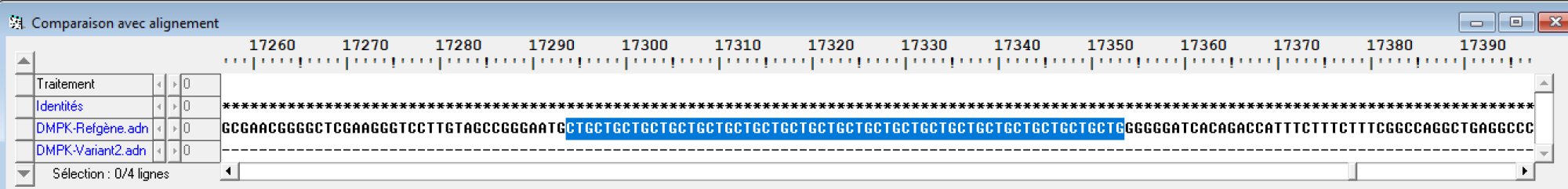
Position des répétitions avec Anagène
On se place (on clique) au début de la séquence. Ensuite, Fichier, Rechercher. On rentre quelques répétitions CTGCTGCTGCTGCTG. Résultat :
3. Les mutations du gène DMPK à l’origine d’allèles causant la DM1
Une répétition comprise entre 5 et 37 triplets n’a pas de conséquences sur l’expression du gène qui est parfaitement fonctionnel. Au-delà de 50 triplets le gène engendre la DM1. Les manifestations de la maladie sont d’autant plus précoces et sévères que le nombre de triplets est élevé. Entre 37 et 50 triplets, on parle de prémutation.
4. Interprétation d’un arbre géologique et estimation d’un risque génétique
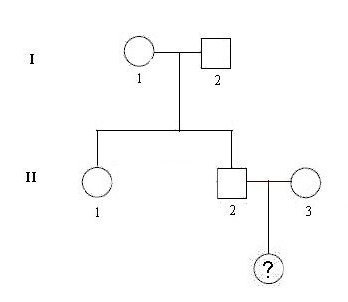
On fournit un arbre généalogique conforme aux données scientifiques.
Les individus de la première génération n’ont pas présenté de symptômes de la DM1. Ils ont eu deux enfants : un fils âgé de 25 ans qui vient d’être diagnostiqué comme présentant les premiers signes de la maladie. Ce fils a comme conjointe une femme au phénotype sans manifestation de la DM1 et le couple attend un enfant Ils se demandent si cet enfant risque d’être atteint par la maladie au cours de sa vie.
Arbre famille DM1
Fichiers des génotypes
Génotype famille DM1.edi (Début au TGA)
Génotype famille DM1-New. edi (Début une vingtaine de pb avant le TGA)
Début d'un allèle

Répétitions dans un allèle

L’exploitation de ces informations génétiques doit conduire à expliquer les génotypes de II2 et II3 à partir de ceux de leurs parents.
La seconde question est d’indiquer la probabilité qu’à l’enfant du couple II2-II3 d’être atteint par la maladie au cours de sa vie.
Arbre famille initiale avec le nombre de CTG.
Suite du dossier à venir...
Références :