
Données et questionnements
Données et questionnements
Première partie
La maladie de Steinert et les notions sur la transmission des maladies héréditaires monogéniques
Ce dossier s’appuie essentiellement sur l’analyse de la maladie de Steinert mais fait aussi appel à d’autres maladies héréditaires monogéniques de façon à avoir une vue d’ensemble des notions de génétique enseignées au lycée.
On utilise couramment l’expression « transmission d‘une maladie héréditaire » mais en réalité ce sont les allèles des gènes qui causent ces maladie qui sont transmis. Néanmoins, dans un premier temps, l’analyse des arbres généalogiques revient à raisonner sur les phénotypes des membres d’une famille pour faire des propositions sur les caractéristiques des gènes impliqués.
I - Diversité des modalités de la transmission des maladies héréditaires monogéniques
A. Cas de la maladie de Steinert
La maladie de Steinert aussi désignée par « dystrophie myotonique de type 1 (en abrégé DM1) est une maladie héréditaire monogénique causée par des allèles d’un gène nommé DMPK (Dystrophy Myotonic Protein Kinase). Elle affecte principalement les muscles entraînant un affaiblissement musculaire progressif (dystrophie) et une difficulté au relâchement après la contraction (myotonie).
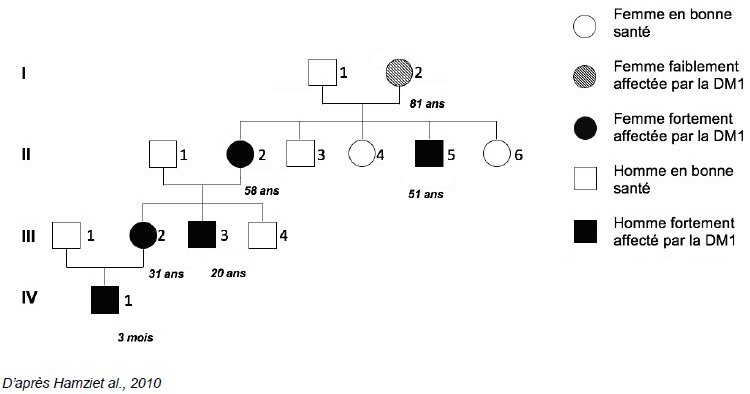
- Document 1 : arbres généalogiques de familles où se transmet la maladie de Steinert
Le premier arbre est relatif à une famille avec indication de l’âge d’apparition des premiers symptômes.
Le deuxième arbre se trouve dans la thèse d’Antoine Mérien « Etude de la fonction des protéines MBNL au cours du développement à l’aide de cellules souches humaines induites à la pluripotence. »
- Document 2 : variabilité du phénotype DM1
C’est sur la base de l’âge d’apparition des premiers symptômes de la maladie, des symptômes cliniques et de leur degré de sévérité que les médecins reconnaissent plusieurs phénotypes, formes de la DM1.
- La forme congénitale, à début néonatal, est la forme la plus grave. Les principaux symptômes sont une grande faiblesse musculaire généralisée et une grande détresse respiratoire.
- La forme juvénile - infantile où les premiers symptômes à se développer sont plutôt d’ordre cognitif avec des difficultés d’apprentissage ; les atteintes musculaires apparaissent généralement à l’adolescence.
- La forme adulte, forme la plus répandue de la DM1, se déclare entre 20 et 40 ans et concerne aussi bien les muscles que les systèmes cardio-vasculaire, respiratoire, endocrinien.
- La forme, tardive la moins sévère de la maladie, avec des atteintes musculaires légères et souvent des problèmes oculaire (cataracte).
Ces différences sont dues à des mutations du même gène DMPK .
Questionnement
A partir des informations fournies par le premier arbre généalogique :
- Indiquer si le phénotype DM1 est dominant ou récessif
- Indiquer un caractère remarquable de la maladie au cours des générations successives
- Montrer que le gène DMPK n’est pas situé sur un chromosome sexuel X ou Y
- En désignant par M+ tout allèle muté du gène DMPK cause de la maladie et par m tout allèle « sain », indiquer le génotype possible des individus malades ;
- Indiquer si le deuxième arbre généalogique confirme les conclusions du premier.
B - Diversité de la transmission des maladies héréditaires monogéniques.
Les dossiers du site ACCES permettent d’étudier la transmission de trois maladies héréditaires : la mucoviscidose, la myopathie de Duchenne (DMD) et ici la maladie de Steinert, dont les modalités de transmission au cours des générations successives sont différentes.
{kind=link}
{kind=link}
Questionnement
Choisir pour chacune de ces maladies une des trois expressions suivantes :
- maladie autosomale récessive
- maladie autosomale dominante
- maladie liée à un chromosome sexuel récessive
- Indiquer pour chacune de ces maladies, au moins un critère de l’arbre généalogique qui lui est propre
Voici un arbre généalogique d’une maladie héréditaire monogénique.
-Indiquer s’il correspond à une des trois maladies précédentes
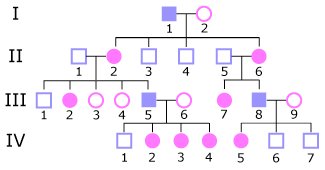
- Indiquer comment on pourrait qualifier la modalité de transmission de la maladie illustrée par cet arbre généalogique
C - Une modalité différente de transmission de maladies héréditaires
Le syndrome NARP est une maladie génétique qui implique une enzyme, l’ATP synthase, localisée dans la membrane interne des mitochondries. Ce syndrome est dû à une mutation du gène MTATP6.
- Document 1
Les deux arbres généalogiques suivants sont relatifs à deux familles dont certains membres souffrent du syndrome NARP.
- Première famille
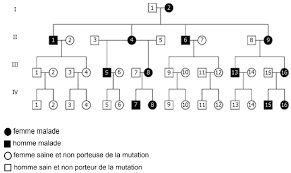
D’après Sujet du bac session septembre 2022 Métropole.
Source : d’après CHU d’Angers – J. Cassereau – Département de neurologie
- Deuxième famille
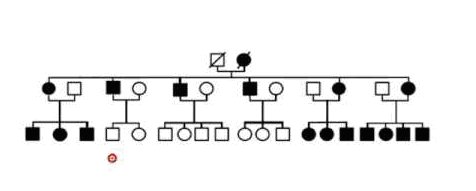
La maladie héréditaire est « Rachitisme vitamino -dépendant » (désignée aussi par hypophosphatémie).
- Document 2 : génétique et comportement des mitochondries au cours de la fécondation
Le noyau n’est pas le seul endroit de la cellule où est localisé l’ADN cellulaire. Les mitochondries possèdent de nombreuses molécules d’ADN où sont localisés quelques gènes.
A la fécondation, l’ovule et le spermatozoïde apportent chacun un lot de mitochondries, mais celles du spermatozoïde sont éliminées et ne contribuent pas à la constitution du patrimoine mitochondrial du zygote (et des cellules de l’organisme qui en dérivent).
Questionnement
- Montrer que chacune des 4 modalités de transmission des maladies monogéniques ne peut expliquer la transmission du phénotype « syndrome de NARP » au sein de la première famille
- Indiquer la caractéristique la plus frappante de l’arbre de la deuxième famille. L’Interpréter en utilisant les informations du document 2
- Justifier le qualitatif « d’hérédité cytoplasmique » donné à ce type d’hérédité. Quel autre qualitatif pourrait-on donner ?
Tests de connaissances et de raisonnements à l’aide de QCM
Pour chaque item, dire pour chaque affirmation si elle est juste ou fausse.
1 - Cas d’une maladie autosomale récessive
- Les hommes malades sont hétérozygotes
- Dans un arbre généalogique, il y a des personnes malades à chaque génération
- Les parents d’une personne malade sont l’un homozygote sain, l’autre hétérozygote
- Une personne malade a toujours deux allèles mutés identiques
2 - Cas d’une maladie liée au chromosome X récessive
- La question de la dominance ou de la récessivité ne se pose pas chez les individus de sexe masculin
- La transmission de la maladie est différente suivant que le parent atteint est le père ou la mère
- Les individus atteints sont surtout des hommes
- Une femme hétérozygote est dite conductrice et transmet l’allèle « sain » à tous ses fils
3 - Cas d’une maladie autosomale dominante
- un seul allèle muté suffit pour que la maladie se manifeste
- une personne malade a un de ses deux parents atteint
- dans un arbre généalogique, on trouve généralement des malades à chaque génération
- très souvent les personnes malades sont hétérozygotes.
4 - Cas d’une maladie dominante liée au chromosome X
- La question de la dominance ou de la récessivité ne se pose que chez les personnes de sexe féminin
- Il y a toujours une transmission père fils
- Toutes les filles d’un homme malade sont atteintes.
- Tous les enfants d’une femme malade sont atteints par la maladie
5 - Dans une maladie mitochondriale
- Les hommes n’ont jamais cette maladie héréditaire
- Une personne malade a sa mère malade
- Les hommes malades ne transmettent la maladie à aucun de leurs enfants
- Les femmes malades transmettent la maladie à tous leurs enfants quel que soit leur sexe.
- La maladie touche les hommes et les femmes de façon comparable.
Deuxième partie
Le gène DMPK , un gène d’eucaryote avec des caractéristiques qui lui sont propres.
A - Recherche de la structure du gène DMPK.
- Document 1 : Rappel sur les étapes de l’expression d’un gène d’eucaryote
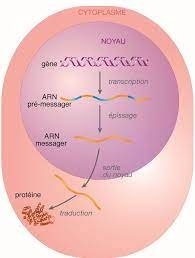
- Document 2 : Séquences relatives au gène DMPK (en ADN) :
Fichier : DMPK-Nouveau.edi
- Séquence de l’allèle de référence du gène DMPK : variant2
Séquence de l’ARN- pré messager du gène DMPK. Remplacer T par U
Séquence de l’ARN messager du gène DMPK. Remplacer T par U
Séquence du CDS du gène DMPK
Séquence de la région 5’UTR du gène DMPK
Séquences de la région 3’ UTR du gène
La séquence codante (CDS) est la région de l’ARN messager qui est traduite en séquence protéique. Elle débute au premier codon AUG de l’ARN messager et se termine au premier codon stop.
Les régions UTR (Untranslated Region) ne sont pas traduites et ne sont donc pas codantes. La première 5’UTR se trouve en amont du codon AUG de début de la traduction. La seconde 3’ UTR va du codon stop à la fin de l’ARN messager. L’ARN polymérase cesse la transcription du gène DMPK lorsqu’elle rencontre dans la région 3’UTR un signal (séquence AATAAA). La fin de l’ARN pré-messager est aussi celle de la séquence de l’ARN messager.
Questionnement
- La transcription du gène DMPK s’effectue t-elle comme celle de tous les gènes eucaryotes ?
- Comment la figure traduit-elle que le gène DMPK est un gène morcelé avec exons et introns ?
- Comment la séquence de l’ARN messager par rapport à celle de l’ARN pré- messager résulte-t-elle de l’épissage ?
- Dans la séquence 3’UTR du gène on trouve le triplet CTG. Indiquer une caractéristique de cette séquence 3’UTR. Comment se retrouve-t-elle dans l’ARN messager ?
- Schématiser la structure de la séquence du gène DMPK
B - Les différences dans les séquences du gène DMPK
Pour les identifier, on dispose des documents suivants :
- Document 1 : Séquences du CDS et de la région 3’UTR de deux allèles. L’allèle A1 est présent en double exemplaire chez une personne qui ne souffre pas de la maladie de Steinert.
L’allèle A2 existe en un seul exemplaire chez une personne présentant des symptômes nets de la maladie de Steinert
- Document 2 Nombre de répétitions CTG et formes de la maladie
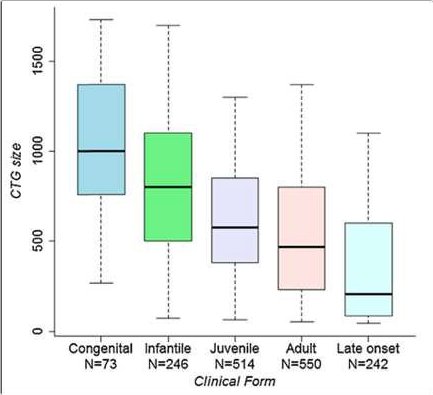
- Document 3 : Nombre de répétitions du triplet CTG au sein d’une famille
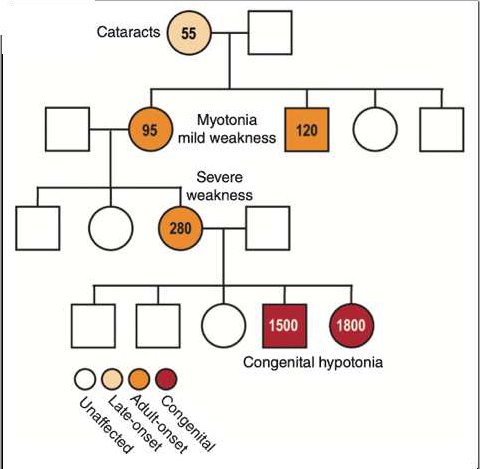
- Document 4 : Schéma bilan sur le gène DMPK
Il faudrait pouvoir changer (CUG)n en (CTG)n
Questionnement
- Est-il exact de dire que la séquence codante d’un allèle DMPK, cause de la maladie de Steinert, diffère de celle d’un allèle « sain » par une mutation ponctuelle (substitution, insertion , délétion) ?
- Les protéines DMPK codées par des allèles différents du gène ont-elles une grande variabilité dan leur séquence ?
- Indiquer en quoi les séquences des allèles du gène DMPK diffèrent
- Le document 3 met l’accent sur le phénomène dit d’anticipation. En quoi consiste t-il ?
- Indiquer ce qui permet de dire que les mutations germinales de Novo sont fréquentes chez les personnes atteintes de la maladie de Steinert. En est-il de même chez les personnes « saines » ? Pourquoi chez les personnes qui ont dans leur séquence du gène DMPK entre 37 et 50 triplets CTG, on parle de prémutation ?
- Indiquer les différentes notions traduites dans le schéma bilan.
Troisème partie
Mécanismes moléculaires de la pathogénèse
La DM1 est une maladie génétique causée par des allèles mutants du gène DMPK. Les mutations sont des expansions du nombre de triplets CTG se trouvant dans la région 3’UTR de la séquence du gène. Il s’agit de comprendre, à l’aide des informations tirées de l’analyse des documents, comment une augmentation du nombre de triplets dans la séquence transcrite mais non traduite et donc non codante du gène DMPK engendre la maladie.
Hypothèses
Peu de temps après avoir établi la séquence du gène DMPK en 1992, les chercheurs ont proposé deux hypothèses explicatives de la pathogénicité.
- La première dite « haploinsuffisance » est basée sur le fait que les malades sont hétérozygotes avec un allèle sain et un allèle responsable de la maladie. Elle suppose que l’expression de l’allèle muté est réduite et aboutit à une production réduite de la protéine DMPK.
- La deuxième suppose que puisque la mutation n’affecte pas la séquence codante, c’est l’ARN messager qui est en cause dans la genèse de la maladie. C’est l’hypothèse de l’ARN messager toxique.
- Document 1 : Comparaison de la concentration en protéines DMPK de biopsies musculaires chez des personnes « saines » et malades.
Ces concentrations ont été déterminées par densimétrie et exprimées en unités arbitraires.
E1, E2, E3 sont des patients ayant un nombre de triplets différent : E1 moins de 500 ; E2 entre 500 et 1000 ; E3 plus que 1000.
|
Données |
Concentration protéines |
pourcentage |
Valeur médiane |
|
Contrôles |
3,62 +-0,69 |
100 |
3,1 |
|
DM1 (E1) |
1,80 +- 0,39
|
50 |
1,9 |
|
DM1 (E2) |
2,11 +-0,43 |
58 |
2,4 |
|
DM1 (E3) |
1,89 +-0,48 |
52 |
2,0 |
|
DM1 (E1+E2+E3 ) |
1,96 =- 0,24 |
54 |
2,1 |
D’après S.Salvatori et al. Decreased expression of DMPK : Correlation with CTG repeat expansionin myotonic dystrophy type 1 Neurol Science 2005 26.
- Document 2 : Expression des deux allèles du génotype d’un patient, l’un « sain », l’autre muté (avec expansion du triplet CTG ). AFMTELETHON juin 2020.
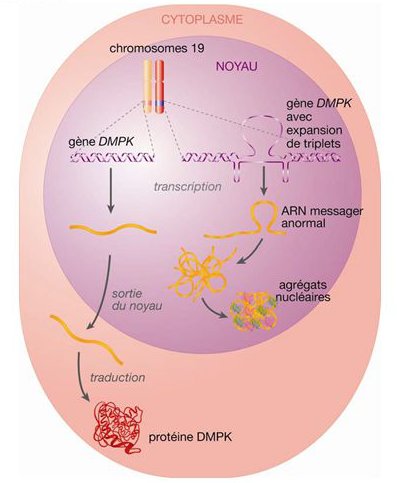
S’il est possible de supprimer la légende : ARN pré-messager ; ARN messager protéines, transcription, épissage, sortie du noyau, traduction ainsi que le 1,2,3 et 4 de la figure ce serait bien (pour poser une question d’exploitation du document.
- Document 3 : Transcrits (ARNm) de l’allèle muté et protéine MBNL
3a. Rôle des protéines MBNL
Les protéines MNBL sont des protéines nucléaires (dans le noyau ) qui interviennent dans l’épissage des ARN pré messagers de nombreux gènes et sont donc indispensables à une expression de ces gènes
3b. Structure en épingles à cheveux de l’ARN messager au niveau de la région d’expansion du triplet CUG.
Les ARN messagers avec une expansion du nombre de triplets CUG qualifiés d’ARN messager anormaux s’accumulent dans le noyau de la cellule où ils forment des agrégats nucléaires.
3c. Relations ARN messagers - protéines MBNL au niveau des agrégats nucléaires

3d. Concentration des protéines MBNL dans le noyau des cellules de personnes atteintes de la maladie de Steinert et de personnes « saines »
Les chercheurs ont évalué chez 3 personnes atteintes de la DM1 et chez 3 personnes « saines » (Contrôles de l’intensité de la concentration des protéines MBNL1 dans le nucléoplasmes des cellules). Dans chaque cas, l’étude a porté sur 20 cellules. Un point du graphique correspond à la teneur en MBNL1 dans une cellule.

- Document 4 : Bilan des conséquences au niveau cellulaire des mutations DMPK
4a. Vue d’ensemble des mécanismes moléculaires en jeu dans la DM1
Il faut supprimer CDK 12 inhibition et sa flèche noire et ce qui se trouve à droite : réduction et les …..
4b. Des ARN messagers altérés aux symptômes de la DM1
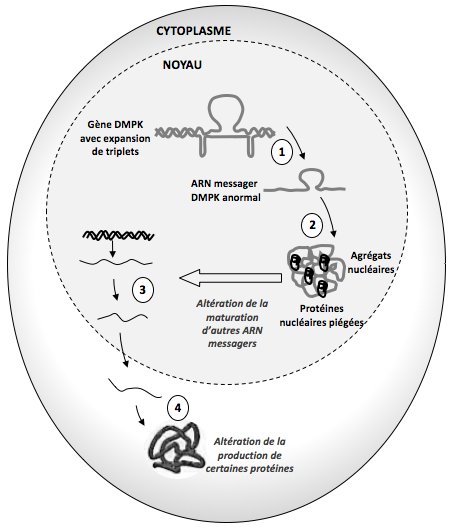
La protéine MBNL1 intervient lors de la maturation de nombreux ARN messagers dont ceux qui permettent de produire :
- Le canal ionique chlore musculaire qui est impliqué dans la myotonie ;
- Le canal ionique sodium cardiaque et la troponine cardiaques qui jouent un rôle dans l’apparition de troubles cardiaques ;
- Le récepteur à l’insuline qui est impliqué dans la résistance à l’insuline ;
- L’amphiphysine et la dystrophine protéines du muscle squelettique dont l’absence provoque son affaiblissaient progressif.
Les déficits en protéines MBNL affectent ces productions.
Questionnements
- - Expliquer en quoi les informations fournies par les documents 1 et 2 confortent l’hypothèse de l’haplo insuffisance.
- - Les protéines DMPK codées par les allèles mutés du gène DMPK ont-elles d’importantes différences dans leurs séquences ?
- - La deuxième hypothèse met en relief la toxicité des ARN messagers mutés du gène DMPK. Résumer les mécanismes à l’origine de cette toxicité.
- - Rédiger un texte qui prend en compte les éléments cités ou représentés dans le premier schéma bilan.
- - Indiquer les phénomènes désignés par les chiffres 1,2,3,4 du deuxième document bilan.
4ème partie
Evaluation d’un risque génétique
Le risque génétique c’est-à-dire le risque de transmission d’une maladie génétique héréditaire correspond à la probabilité de transmettre ou de recevoir l’anomalie génétique et la maladie génétique en cause. Pour cela on s’appuie sur un arbre généalogique qui établit les relations de parenté entre les membres d’une famille , le phénotype clinique de chacun d’entre eux, et leur statut génétique si un test génétique a été réalisé.
A - Risque génétique dans le cas de la maladie de Steinert
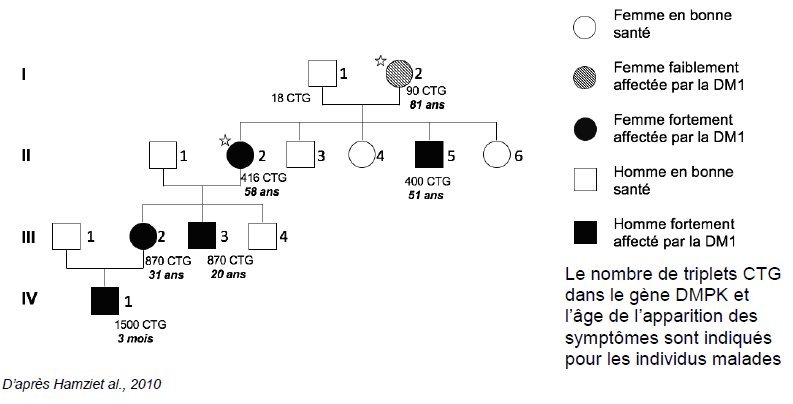
- Document 1 : Arbre généalogique familial.
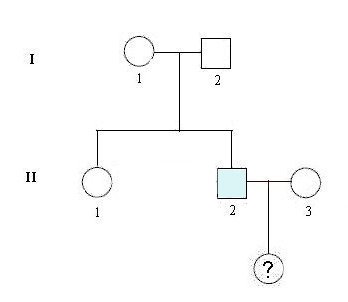
L’Homme II2 âgé de 25 ans vient d’être diagnostiqué comme présentant les premiers signes de la maladie. Le couple II2-II3 attend un enfant et il se demande si cet enfant risque d’être atteint de la maladie au cours de sa vie et avec quelle gravité.
Questionnement
Le risque génétique dans le cas de la maladie de Steinert
- L’arbre généalogique confirme t-il que la maladie de Steinert est une maladie génétique dominante ?
- En utilisant les données sur les génotypes des membres de la famille, expliquer les différences entre les génotypes de II1 et II2
- Proposer une explication au fait que ii2 soit atteint par la maladie de Steinert
- Calculer le risque ‘qu’a l’enfant à naître d’être atteint de la maladie de Steinert durant sa vie. Y a-t-il un risque qu’il soit plus gravement atteint que son père ?
- Document 2 : séquences 3’UTR des allèles du gène DMPK possédés par les individus I1 ; I2 ; II1 ; II2 ; II3.
- Tests de connaissances et raisonnements sur le risque génétique relatif à d’autres maladies héréditaires.
1 - Cas d’une maladie autosomale récessive
Le risque qu’a une soeur hétérozygote saine d’un sujet malade d’avoir à chaque grossesse un enfant atteint de la maladie est de :
- 0% si son conjoint es hétérozygote
- 100% si son conjoint est un hétérozygote sain
- 0% si son conjoint est un homozygote sain
- 25% si son conjoint est un porteur sain
2 - Cas d’une maladie autosomale récessive
La fréquence des hétérozygotes parmi les apparentés d’un sujet malade est de :
- 100% pour les parents
- 2/3 pour les frères et soeurs sains
- 0% si pour les enfants qu’a le malade avec un hétérozygote sain
- 1/3 pour les neveux et nièces
3. Cas d’une maladie autosomale récessive
La phénylcétonurie est une maladie héréditaire où la prévalence à la naissance est de 1/10000 (un enfant sur 10000 nait avec la phénylcétonurie). Cela correspond à une fréquence de porteurs sains de 2% dans la population
Considérons le cas d’un couple de personnes non apparentées ; l’homme a un frère atteint de phénylcétonurie alors que la famille de sa conjointe on ne connaît pas de cas de phénylcétonurie.
Le risque qu’a ce couple d’avoir un enfant malade est de :
a) 50%
b) 0,2%
c) 1/300
d) 1/10000
4. Cas d’une maladie autosomale dominante
Considérons le cas d’un homme malade mais hétérozygote,
Le risque d’avoir un enfant malade est de :
- 50% si sa conjointe n’est pas atteinte par la maladie
- 100% si sa conjointe est atteinte par la maladie
- Il peut avoir un frère malade et une soeur saine
- Le risque que sa soeur ait chaque grossesse un enfant malade est de 0% que son conjoint soit malade ou non
5. Cas d’une maladie dominante liée au chromosome X
Le risque de transmettre la maladie est :
- Le même que le parent atteint soit le père ou la mère
- Dans le cas d’une mère malade et d’un père sain, le risque à chaque grossesse d’avoir un garçon malade ou une fille malade est le même
- Dans le cas d’un père malade et d’une mère saine, a chaque grossesse, le risque qu’un garçon soit malade est de 50% et celui qu’une fille le soit est de 100%
- Si le père et la mère sont malades, à chaque grossesse, le risque que l’enfant le soit aussi est de 100%
6. Cas d’une maladie récessive liée au chromosome X
L’hémophilie est due à un défaut de la coagulation du sang causée par des allèles d’un gène situé sur le chromosome X :
- La fréquence de la maladie est la même chez les garçons et les filles
- La transmission de l’hémophilie est différente suivant que le parent atteint soit le père ou la mère
- Si le père est hémophile et la mère porteuse saine (= conductrice), tous les garçons auront un risque sur deux d’être hémophiles et toutes le filles seront non hémophiles mais porteuses saines (= conductrices)
- Le risque qu’un père hémophile transmette la maladie à ses fils est nul.