
De l'identification de la cause moléculaire d'un cancer à son traitement
De la découverte progressive de la cause d’une maladie à la mise au point d’un traitement
L’objectif de ce dossier n’est pas la connaissance de la leucémie myéloïde chronique et de son traitement. Cette maladie n’est qu’un support pour illustrer comment l’élucidation progressive des mécanismes en jeu dans une maladie, ici un cancer de cellules sanguines, a pu conduire à un traitement ciblé vis-à-vis de cette maladie. Dans le domaine de la cancérisation, c’est le premier exemple de thérapie ciblée, On montre comment une succession de découvertes de recherche fondamentale, permises par la mise au point de nouvelles technologies, a débouché sur une recherche appliquée ayant conduit à un traitement améliorant beaucoup le pronostic de la maladie. C’est aussi l’occasion pour des élèves de première de réinvestir les concepts de base de biologie cellulaire, de biologie moléculaire.
1 - Les caractéristiques de la leucémie myéloïde chronique
Le document : « La leucémie myéloïde chronique » n’est pas vraiment un document de travail. Il fournit les informations minimales pour exploiter les autres documents. Il s’appuie sur les connaissances vues en troisième sur les cellules sanguines, notamment les globules blancs. Les clichés sur les frottis sanguins permettent de visualiser les anomalies, liées à la LMC pendant la phase chronique et la phase aigüe. Le frottis relatif à la phase chronique est légendé de façon à servir de référence et de permettre de légender les deux autres frottis.
Les promyélocytes et les métamyélocytes sont des précurseurs de polynucléaires n’ayant pas totalement achevé leur différenciation. Ils ne sont pas présents dans un frottis sanguin normal ; ils peuvent être absents d’une LMC au début de sa phase chronique. Dans le frottis sanguin en phase aiguë de la maladie, on voit beaucoup de grosses cellules qui sont des leucocytes immatures (Myéloblastes).
2 - La découverte d’une anomalie chromosomique associée à la maladie
- L’idée que des anomalies chromosomiques peuvent être en cause dans la cancérisation des cellules est ancienne, formulée notamment par Boveri en 1914. Mais cette idée a été laissée de côté pendant plusieurs décades faute de disposer de techniques permettant une observation précise des chromosomes des cellules. Les années 50 voient une avancée importante dans les techniques de la cytologie avec la culture des tissus, l’utilisation de la colchicine qui bloque les mitoses en métaphase, et l’utilisation de solutions hypotoniques pour entraîner un gonflement des cellules, étape indispensable à l’obtention d’un étalement des chromosomes. Grâce à l’utilisation de ces techniques, Tijo et Levan établissent en 1956 que le nombre de chromosomes humains est de 46, et Turpin, Gautier et Lejeune, que les personnes atteintes du syndrome de Down ont 47 chromosomes par suite de la présence d’un petit chromosome supplémentaire. Une nomenclature internationale aboutissant à une classification telle qu’elle apparaît actuellement dans les caryotypes est établie à Denver en 1960. Le chromosome supplémentaire lié au syndrome de Down est identifié comme étant le chromosome 21.
- C’est dans ce climat de découvertes dans le domaine de la cytologie, que deux chercheurs de l’université de Pennsylvanie Nowell .et Hungerford, travaillant sur les cancers, et plus particulièrement sur les leucémies, ont utilisé les nouvelles techniques pour tester l’idée de Boveri. Ils ont fait la culture de cellules de la moelle osseuse de 7 patients atteints de LMC et analysé leur caryotype. Ils ont trouvé dans tous les cas la présence d’un petit chromosome, absent dans un caryotype normal. Dans l’article de Science de 1960 relatant leurs travaux, ils disent : nous avons observé un petit chromosome remplaçant un des 4 plus petits chromosomes du caryotype humain. Tous les 7 individus montraient le même petit chromosome. Et aucune autre anomalie n’a été constatée. En outre le caryotype des cellules non cancéreuses de ces patients était normal ne montrant pas ce petit chromosome. Ainsi le petit chromosome était spécifique d’un type bien défini de cancer : la LMC. Ce chromosome a été appelé chromosome Philadelphie (Ph) du nom de la ville où la recherche avait été faite. Cette corrélation ne prouvait pas que ce chromosome était le facteur causal de la LMC ; peut-être en était-il seulement une conséquence.
- Si les travaux des années 60 ont confirmé la liaison chromosome Ph-LMC, il a fallu attendre les recherches de Janet Rowley en 1973 pour comprendre l’origine de ce chromosome Ph. Elle a a eu recours à de nouvelles techniques d’observation des chromosomes, notamment l’utilisation du Giemsa et de la quinacrine. Ces techniques mettent en évidence une structure plus fine des chromosomes notamment l'existence de bandes spécifiques de chaque chromosome. Cette étude a révélé que le chromosome Ph résultait d’une translocation réciproque entre les chromosomes 9 et 22. Le chromosome Ph était un chromosome 22 sans une grande partie de son bras long, mais avec la partie terminale du bras long du chromosome 9. Les individus atteints de LMC avaient aussi un autre chromosome anormal non repéré par Nowell et Hungerford, le chromosome 9. C’était donc une double translocation qui était associée à la LMC.
- Dans leur article de Science, Nowell et Hungerford ne fournissent pas de cliché et les photos présentes dans l’article de Rowley sont trop difficiles à interpréter par des élèves de première. Les figures du document « Une anomalie chromosomique liée à la LMC » sont extraites d’articles de seconde main sur la LMC ; tout en télescopant certaines étapes de la démarche historique elles en respectent globalement l’esprit.
- Le travail avec les élèves peut commencer par une comparaison des caryotypes des cellules normales et cancéreuses afin de rechercher une ou des caractéristiques du caryotype des cellules cancéreuses. La première conclusion doit être qu’il n’existe pas de différence dans le nombre de chromosomes des deux types de cellules. La deuxième conclusion est le repérage des paires de chromosomes où les deux chromosomes présentent des différences entre cellules normales et cellules cancéreuses : les paires 9 et 22. Dans les cellules normales, les deux chromosomes de ces deux paires ont la même longueur alors que pour les cellules cancéreuses un des chromosomes 9 est plus long qu’un chromosome 9 normal et un des chromosomes 22 plus petit qu’un chromosome 22 normal.
- Le gros plan sur les paires de chromosomes 9 et 22 dans une cellule leucémique LMC confirme les conclusions de l’analyse des caryotypes ; un des chromosomes 9 possède à une de ses extrémités un matériel supplémentaire par rapport à un chromosome 9 normal, et un des chromosomes 22, le chromosome Ph, a une extrémité raccourcie par rapport au chromosome normal. Les élèves peuvent proposer diverses explications pour rendre compte de ces données, notamment la délétion d’une partie du chromosome 22, l'addition d’un segment sur le chromosome 9. En réfléchissant sur ce qu’est devenu le segment délété du chromosome 22 et sur l’origine du segment ajouté au chromosome 9, ils peuvent imaginer une translocation d’une partie du chromosome 22 sur le chromosome 9.
- La schématisation de la structure en bandes des chromosomes des paires 9 et 22 d’une cellule leucémique permet de tester ces hypothèses et de s’apercevoir qu’aucune n’est tout à fait exacte.
On s’aperçoit que l’organisation en bandes du bras court du chromosome Ph est identique à celle d’un chromosome 22 normal. En revanche celle du bras long Ph est différente de celle du bras long du chromosome 22. Mais elle est identique à celle de l’extrémité du bras long du chromosome 9. Ainsi le chromosome Ph apparaît comme un chromosome chimère ayant une partie du chromosome 22 associée à une partie du chromosome 9. L’évènement ayant conduit à ce chromosome chimère implique une cassure d’une part sur un chromosome 9, d’autre part sur un chromosome 22, suivies d’une translocation de la petite partie terminale du chromosome 9 sur le chromosome 22.
Des observations du même type conduisent à dire que le chromosome 9 anormal des cellules leucémiques est aussi un chromosome chimère ayant la majeure partie d’un chromosome 9 normal associée à la plus grande partie du bras long du chromosome 22. On peut donc parler d’une double translocation et on peut demander aux élèves de la schématiser. Une schématisation possible est la suivante.

D'après NIH.
3 - Les conséquences génétiques de cette translocation réciproque
Le fait que cette translocation réciproque 9-22 soit quasiment présente dans toutes les cellules leucémiques des patients atteints de LMC plaide très fortement en faveur de l’idée que cette translocation est un facteur causal de la leucémie. Elle confirme aussi l’idée que les cellules leucémiques résultent d’une mutation chromosomique survenue dans une cellule souche initiale et transmise ensuite à toutes ses descendantes.
Pour confirmer cette idée il fallait découvrir les changements moléculaires dus à la translocation susceptibles de cancériser les cellules. Cela fut élucidé durant les années 80.
Le point de départ a été la mise en évidence de l’oncogène présent dans un rétrovirus (rétrovirus Abelson) provoquant une leucémie chez la souris. Les chercheurs ont ensuite trouvé l’homologue de cet oncogène viral dans le génome des cellules normales, c'est-à-dire un proto oncogène.
Dès 1982 ils ont localisé le proto oncogène ABL (symbole dérivé du virus) sur le chromosome 9 chez l’homme et précisément dans une région qui est transloquée dans les cellules leucémiques.
De même ils ont localisé sur le chromosome 22 une région appelée BCR où avait lieu la rupture préalable à la translocation du bras long du chromosome 22 sur le chromosome 9. Par la suite cette région s’est révélée correspondre à un gène qu’on a appelé BCR.
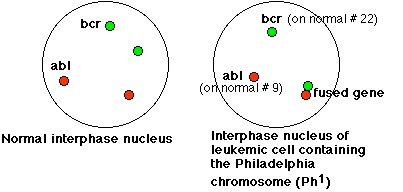
Sachant que le gène ABL du chromosome 9 était un proto oncogène, les chercheurs ont voulu déterminer si ce gène était transloqué sur le chromosome 22 au cours de la formation du chromosome Philadelphie. Le cliché d’hybridation in situ du document : Une conséquence génétique (moléculaire) de l’anomalie chromosomique met en évidence les gènes BCR et ABL dans une cellule normale et dans 5 cellules leucémiques d’un patient atteint de LMC.
Cette hybridation a été réalisée sur des cellules interphasiques, ce qui explique qu’on ne distingue pas les chromosomes des noyaux. Dans une cellule normale, on voit deux tâches vertes qui correspondent chacune à un allèle du gène BCR situé sur un des chromosomes 22. De même, on voit deux tâches rouges qui correspondent chacune à un allèle du gène ABL situé sur un des chromosomes 9. On peut demander de schématiser la localisation des allèles de chacun de ces gènes sur les chromosomes.
Dans toutes les cellules leucémiques, on distingue une tâche verte et une tâche rouge qui correspondent respectivement à un allèle du gène BCR et à un allèle du gène ABL. En outre, on distingue dans chaque cellule leucémique une tâche rouge associée à une tâche verte. Cela correspond à un allèle du gène BCR associé à un allèle du gène ABL sur un même chromosome, le chromosome 22. Le schéma ci-dessous est une interprétation des figures d’hybridation
D'après : Chronic Myelogenous Leukemia (CML)
Le schéma sur la localisation chromosomique des allèles BCR et ABL, conclusion de cette expérience d’hybridation, traduit bien que le gène ABL a été transloqué du chromosome 9 au chromosome 22. A ce stade, on ne peut pas vraiment parler de fusion entre les deux gènes, ils apparaissent comme juxtaposés sur le chromosome Philadelphie.
Les informations fournies par les séquences
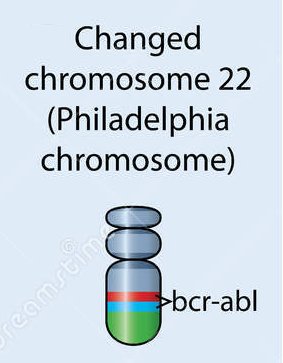
En 1984, on a identifié dans les cellules leucémiques un ARNm qui code pour une protéine BCR-ABL non présente dans les cellules normales. La comparaison avec Anagène de la séquence de cette protéine avec celle de la séquence de la protéine BCR puis avec celle de la protéine ABL d’une cellule normale révèle que la protéine BCR-ABL est une protéine chimère dont la région amont correspond à celle de la région amont de la protéine BCR et la région suivante à celle de la région aval de la protéine ABL. Le dotplot visualise bien cette constitution chimérique de la protéine BCR-ABL.

Cela signifie que le gène codant pour cette protéine est aussi un gène chimère. Autrement dit, les gènes BCR et ABL ont été coupés au cours de la translocation et il en est résulté un gène de fusion au lieu de deux gènes, gène qu’on désigne aussi par BCR-ABL. C’est ce que traduit le schéma sur le chromosome Philadelphie du document.
Il restait à déterminer si la protéine codée par ce gène de fusion, engendré par la translocation, était bien responsable de la cancérisation de la cellule.
Une confirmation expérimentale de l’action oncogénique de la protéine BCR-ABL
Cette expérience de transgénèse conduit à des souris qui ont intégré dans le génome de leurs cellules souches de la moelle osseuse, le transgène BCR-ABL humain. Les souris donneuses et receveuses sont de même lignée, ce qui évite un rejet des cellules injectées par les receveuses. Quelques semaines après l’injection, un frottis sanguin réalisé chez les souris receveuses montre une richesse en globules blancs, en particulier en polynucléaires nettement supérieure à la normale. Dans leur article, les auteurs indiquent 15000 à 500000 leucocytes par mm3 alors que le nombre normal est de 5000 par mm3. D’autre part la rate est hypertrophiée.
Ce sont là des données qui évoquent celles observées chez un patient humain atteint de LMC au cours de la phase chronique. Cela n’est pas observé chez des souris irradiées ayant reçu des cellules souches n’ayant pas incorporé le transgène BCR-ABL. On peut donc conclure que la protéine codée par le transgène a la propriété de déclencher une multiplication anormale des cellules souches granuleuses de la moelle osseuse.
4 - Action des protéines ABL et BCR-ABL
Les 2 protéines ABL et BCR-ABL ont la même fonction kinasique qui se traduit finalement par l'activation des protéines stimulant la prolifération cellulaire. Cette similitude d'action s'explique, comme l'a montrée l'analyse des séquences par le fait que la protéine BCR-ABL comprend la majeure partie de la protéine ABL, en particulier la région responsable de l'action enzymatique. Mais la séquence de BCR qui figure en amont de la séquence ABL dans la protéine BCR-ABL fait que celle-ci est constamment active et stimule donc en permanence la prolifération cellulaire ; elle est donc oncogénique. Inversement, dans les cellules souches normales, la protéine ABL est soumise à régulation et ne devient active qu'en fonction de signaux reçus par les cellules de leur environnement.
Le gène ABL est un proto-oncogène, et le gène BCR-ABL un oncogène.
5 - Une thérapie ciblée : le Glivec (ou Gleevec) ou Imanitib
La recherche fondamentale sur la LMC de 1960 à 1990, associée aux progrès techniques en biologie cellulaire (cytologie) puis moléculaire, ont conduit à la conclusion que la double translocation entre un chromosome 9 et un chromosome 22, survenue dans une cellule souche sanguine, était à l’origine de la LMC. Elle aboutit à la création sur le chromosome 22 modifié (chromosome Philadelphie) d’un gène de fusion codant pour une protéine de fusion BCR-ABL. Cette protéine suffit pour transformer une cellule souche. L'élucidation du mécanisme moléculaire à l’origine de la LMC fournissait une piste pour un traitement de la LMC : il fallait trouver une substance capable d’inhiber l’action de la protéine BCR-ABL. C’est le premier exemple d’une recherche appliquée en cancérologie ciblée sur un agent causal de la cancérisation d’une cellule. A ce titre la LMC est spéciale parmi les cancers du fait de l’unicité de sa cause initiale.
Pour cette recherche appliquée, les chercheurs se sont basés sur le mode d’action de la protéine BCR-ABL à savoir que c’est une protéine kinase activant spécifiquement des protéines cellulaires qui stimulent la prolifération des cellules. Il fallait trouver une substance susceptible d’empêcher le transfert d’un groupement phosphate de l’ATP sur ces protéines cibles. Vers la fin des années 90, une substance, l’imatinib ou Glivec , a été synthétisée qui s’est avérée active lors d’essais précliniques.
Le document « Une thérapie ciblée : le Glivec » schématise l’action du Glivec.
On y voit que, normalement, l’ATP se fixe dans une « poche » de la structure tridimensionnelle de la protéine BCR-ABL, ce qui est indispensable pour que cette enzyme transfère un groupement phosphate sur une protéine substrat. En raison d’une conformation spatiale proche de l’ATP, la molécule d’imatinib prend la place de l’ATP dans cette poche. Ainsi, l’activité de l’enzyme est bloquée. Les protéines substrats ne sont pas phosphorylées et restent inactives. La prolifération cellulaire n’est plus stimulée. Il s’agit d’une inhibition de l’activité enzymatique par compétition.
Des essais cliniques ont été autorisés dès 2000 et les graphiques du document : « Une thérapie ciblée le Glivec » illustrent les résultats d’un essai clinique sur 5 ans de 2000 à 2005. Il a été fait sur 553 personnes chez qui on a diagnostiqué une Leucémie myéloïde chronique et qui ont été traités par une prise journalière de Glivec (400 mg par jour). Pour bien apprécier la signification des résultats obtenus, il faut avoir à l'esprit qu’en l’absence de traitement, les patients entrent dans une phase de leucémie aigüe (blastique) mortelle 4 à 6 ans après le diagnostic de Leucémie myéloïde chronique.
Le premier graphique indique que 5% environ des patients traités par le Glivec sont entrés en phase aigüe de leucémie mortelle. Donc, 95% des patients ont survécu. C'est très nettement supérieur à la mortalité sur 5 ans sans traitement et illustre donc l'efficacité du Glivec.
Le deuxième graphique, permet d'affiner les réponses des patients au Glivec et de faire percevoir une certaine variabilité.
On constate ainsi que parmi les patients ayant survécu à 5 ans, 96% ont eu une réponse hématologique complète à 1 an, et 98% à 5 ans. Cela signifie que la formule sanguine de ces patients est redevenue normale et qu’en particulier il n’y avait plus d’hyperleucocytose. Le rythme de multiplication des cellules souches à l’origine de la leucémie a donc diminué, ce qui est une preuve de l’efficacité du Glivec. Cependant, une réponse cytologique complète n’est atteinte que pour 69% des patients à 1 an et 87% à 5 ans. Une réponse cytologique complète implique l’absence dans la moelle osseuse de toute métaphase où un chromosome Philadelphie est repérable. Cela signifie que chez 27% des patients (96 - 69 = 27) à un an, la leucémie n’est plus décelable à l’état clinique mais le reste au niveau cellulaire. Ce nombre n’est plus que de 11% à 5 ans. La courbe sur la réponse cytogénétique majeure montre que chez une partie des patients n’ayant pas une réponse cytogénétique complète, le Glivec a, malgré tout, une action positive avec une réduction des cellules de la moelle osseuse possédant le chromosome Philadelphie.
Depuis, on a constaté chez certains patients traités par le Glivec une rechute, c'est-à-dire un retour des anomalies sanguines caractéristiques de la LMC. On a recherché des explications à ces rechutes et on constaté qu’elles étaient dues à des mutations du gène BCR-ABL modifiant ponctuellement la séquence d’acides aminés de la protéine BCR-ABL dans la région kinasique, notamment, celle qui fixe l’ATP.
La connaissance de ces mutations ponctuelles a servi de guide pour la mise au point de nouveaux médicaments actifs contre la LMC. On a à nouveau une illustration du fait que la recherche fondamentale joue un rôle crucial dans la mise au point de thérapies ciblées.
Les élèves pourraient consulter utilement ce document intitulé : "BCR-ABL: Cancer Protein Structure and Function".