
Mécanismes cellulaires et moléculaires de la drépanocytose
I. Mécanismes cellulaires et moléculaires de la drépanocytose
Comme pour toutes les thérapies géniques, celle relative à la drépanocytose repose sur la connaissance des mécanismes cellulaires et moléculaires impliqués dans la maladie. Ceux-ci sont envisagés sur la plateforme Access dans la thématique biologie moléculaire et évolution. Elle donne accès à un accompagnement pédagogique au lycée sur les phénotypes drépanocytaires en seconde (les phénotypes drépanocytaire et non drépanocytaire et le gène de l’hémoglobine), en première de spécialité (expression de l’information génétique : transcription et traduction) ; mutations et santé : les hémoglobinopathies), en terminale spécialité (famille multigénique des globines).
Dans cette introduction, on résume les notions essentielles qui sont nécessaires pour comprendre les modalités de la thérapie génique de la drépanocytose En outre, on fait appel aux données du modèle de thérapie génique, précédemment envisagée, celle de la thérapie relative au DICS-X.
A - Les causes moléculaires de la drépanocytose
La drépanocytose est une maladie génétique héréditaire causée par une anomalie des molécules d’hémoglobine. Celles-ci sont des tétramères formés par l’association de deux polypeptides alpha et de deux polypeptides bêta. La figure ci-dessous indique de façon un peu simplifiée la localisation des gènes codant pour ces polypeptides. Les gènes bêta sont localisés sur le chromosome 11 et les gènes alpha sur le chromosome 16.
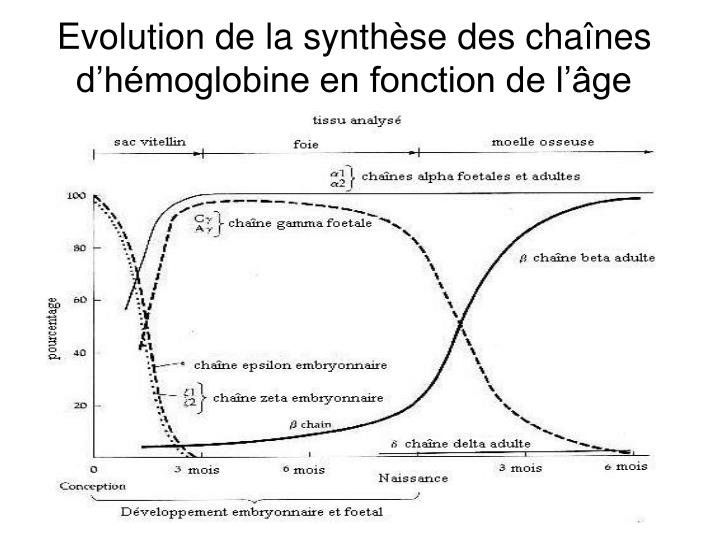
Le graphique ci-dessous renseigne sur l’expression de ces gènes durant la vie embryonnaire et fœtale puis après la naissance.
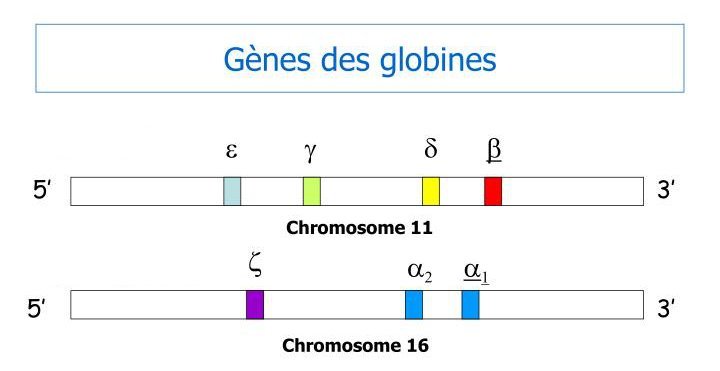
L’hémoglobine foetale HbF est formée durant toute la vie foetale mais sa synthèse s’arrête dans les premiers mois après la naissance. Les tétramères de HbF sont constitués par l’union de deux molécules alpha et de deux molécules gamma. Après la naissance les tétramères de HbA sont synthétisés pendant toute la vie. Ils sont constitués par l’association de deux molécules alpha avec deux molécules bêta. Le point important pour la thérapie génique est l’existence d’un « switch » dans la période qui suit la naissance : arrêt de l’expression du gène gamma remplacée par celle du gène bêta.
La drépanocytose (Sickle cell disease) est causée par une mutation du gène qui code la chaîne bêta de l’hémoglobine HbA. La mutation est une substitution au sixième codon de la séquence codante, qui fait que la valine remplace l’acide glutamique au 6ème acide aminé de la chaîne bêta. La maladie ne se manifeste que chez les personnes homozygotes où les deux gènes bêta sont mutés. On désigne par bêta S l’allèle muté et par HbS l’hémoglobine drépanocytaire. Puisque le gène bêta S ne s’exprime qu’après la naissance, les signes de la maladie n’apparaissent qu’au bout de quelques mois de vie.
Les tétramères HBS se polymérisent lorsque la pression en dioxygène des globules rouges est basse par un mécanisme illustré par la figure suivante.
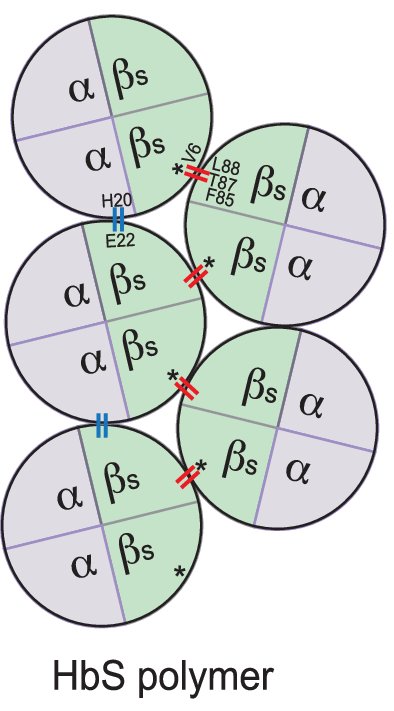
Explication de la polymérisation de HbS : dans les polymères HbS , la valine en position 6 de la chaîne bêta S établit un contact latéral avec la phénylalanine et la leucine aux positions 85 et 88 (F85 et L88) de la chaîne bêta S d’un tétramère adjacent. En plus, un acide glutamique, à la position 22 de la chaîne bêta S, interagit avec une histidine, en position 20 de la globine alpha d’un tétramère voisin (contact axial).
Figure extraite de « Gene Therapy for bêta hémoglobinopaties » Marina Cavazzana, Chiara Antoniani and Annarita Miccio. Mol Ther. 2017 May 3; 25(5): 1142–1154.
La polymérisation des tétramères HbS forme au sein des globules rouges des fibres qui les rendent rigides et prennent une forme en faucille. Cette falciformation est à l’origine des manifestations cliniques de la maladie : anémie, crises vaso-occlusives très ddouloureuses, atteinte de divers organes, sensibilité aux infections.
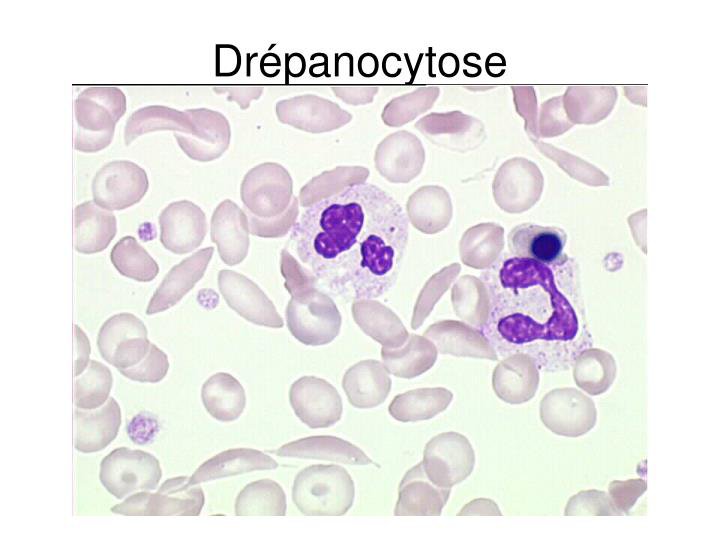
Frottis sanguin d’une personne drépanocytaire
B - L’hémoglobine foetale HbF et la drépanocytose
Certaines personnes de génotype bêta S//bêta S ont des manifestations cliniques de la drépanocytose très modérées. Toutes sont caractérisées par une persistance de l’hémoglobine foetale HbF après la naissance. Les globules rouges de ces personnes possèdent donc des tétramères HbS et aussi des tétramères HbF (10 à 30%). Le gène gamma continue donc à s’exprimer chez elles après la naissance. Ces données cliniques indiquent que, dans une certaine mesure, l’hémoglobine foetale exerce un effet inhibiteur sur la polymérisation de l’hémoglobine HbS. La figure en fournit une explication .
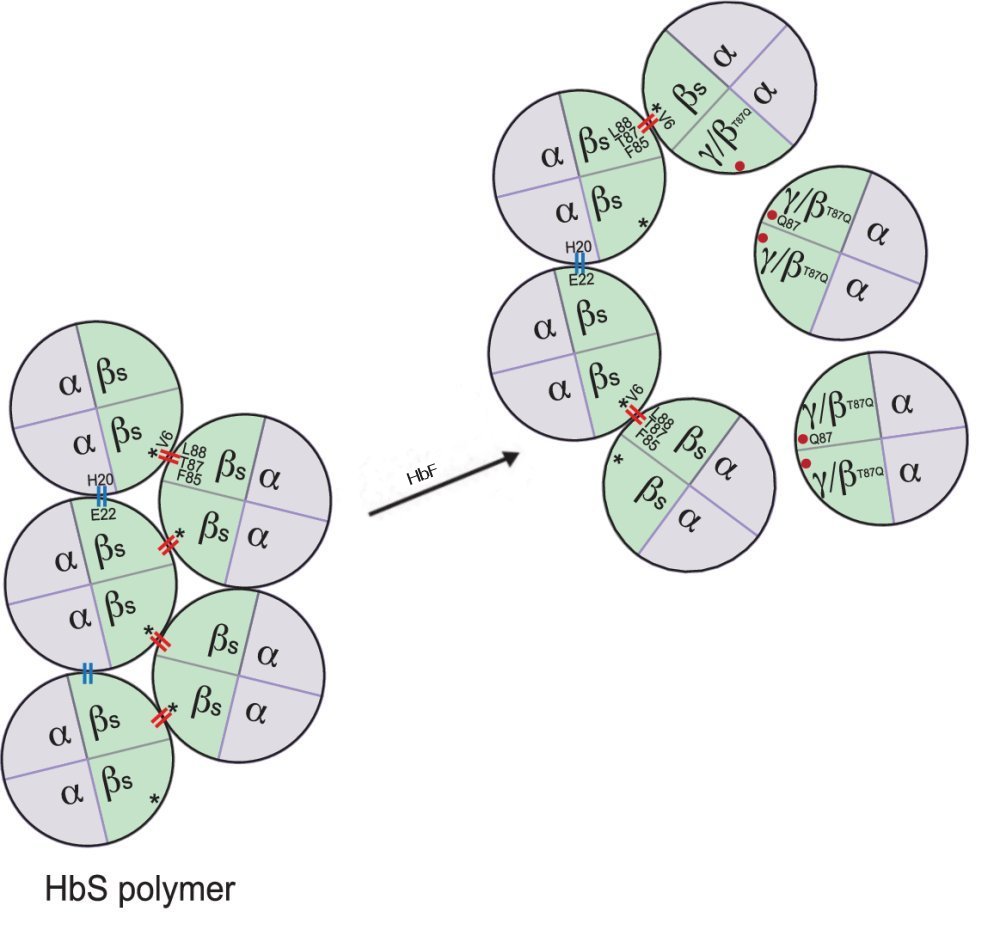
Explication de l’effet inhibiteur de le HbF sur la polymérisation de HbS.
La chaîne gamma possède en position 87, à la place de la thréonine, l’acide aminé glutamine qui inhibe la formation de contacts latéraux entre les tétramères de HbS.
Les tétramères ayant deux monomères où c’est indiqué Gamma/bêta T87Q correspondent à deux situations. La première est celle où le monomère est la chaîne gamma (il s’agit donc de l’hémoglobine foetale). La deuxième est celle où le monomère est une chaîne bêta modifiée(T87Q). Cette chaîne est en jeu dans la thérapie génique envisagée plus loin.