
Le gène RB
Les caractéristiques du rétinoblastome
Cependant, la technique de fusion cellulaire n’a pas débouché sur l’identification de ces gènes suppresseurs de tumeurs. C’est l’étude d’un type rare de cancer, le rétinoblastome, au cours des années 70 et 80 qui devait aboutir au clonage et au séquençage (1987) du premier gène suppresseur de tumeur, le gène RB (ou RB1).
 Document « Les caractéristiques du rétinoblastome »
Document « Les caractéristiques du rétinoblastome »
Le document permet d’abord de dégager les caractéristiques communes à tous les rétinoblastomes : c’est un cancer qui apparait précocement, avant l’âge de 5 ans, et qui se traduit par au moins une tumeur à l’intérieur d’un œil. Cette tumeur a pour origine une cellule de la rétine qui, au cours du développement, au lieu de se différencier (en cellule photo réceptrice par exemple) a conservé un caractère embryonnaire et s’est multipliée de façon incontrôlée.
Les arbres généalogiques permettent de reconnaître deux formes de rétinoblastome. Dans l'une de ces formes, la forme A, on constate la présence de personnes atteintes dans plusieurs générations de la famille (cela suppose que les personnes atteintes aient été bien soignées pour atteindre l’âge adulte). Vu que le rétinoblastome est un cancer rare dont la fréquence est estimée à 1/20.000, cela signifie que le rétinoblastome dans une telle famille est héréditaire. En outre, le fait qu’on trouve une personne atteinte dans chaque génération s’interprète bien en admettant qu’un seul gène est en cause et que l’allèle muté est dominant. A vrai dire, on verra plus loin que ce n’est pas le cas. On peut seulement dire que le phénotype rétinoblastome est dominant.
Dans la forme B, le sujet atteint apparaît au sein d’une famille où on ne connaît pas de cas de rétinoblastome. C’est d’ailleurs la forme la plus fréquente (60% des cas environ). La personne atteinte n’a pas d’individus affectés dans sa descendance. On parle de rétinoblastome sporadique, non héréditaire.
L’arbre généalogique Cest une variante de rétinoblastome héréditaire où il n’y a pas de personnes atteintes dans l’ascendance du premier sujet atteint mais où on en trouve dans la descendance de ce dernier. Il est intéressant de demander aux élèves une explication dans le cadre d’une hypothèse où un seul gène est en cause. Ils doivent imaginer une mutation de novo dans la lignée germinale d’un des parents du premier sujet atteint.
On peut alors faire le point sur les différences phénotypiques entre les deux formes de rétinoblastome. Le rétinoblastome héréditaire apparaît en moyenne plus précocement que le rétinoblastome sporadique.
Enfin le rétinoblastome héréditaire affecte souvent les deux yeux et il peut y avoir plusieurs tumeurs dans le même œil. Comme la tumeur a pour origine une seule cellule, cela signifie que plusieurs cellules sont devenues cancéreuses dans le cas du cancer héréditaire ce qui n’est pas le cas avec le rétinoblastome sporadique.
Ces différences soulèvent des questions ou hypothèses : est-ce que c’est le même gène qui est en cause dans les deux cas ? Si c’est le même gène, on peut supposer que c’est la transmission héréditaire d’au moins un allèle muté qui peut faire que le rétinoblastome héréditaire est d’apparition plus précoce et peut toucher plusieurs cellules rétiniennes.
Le modèle génétique explicatif de Knudson (1971)
C’est à partir d’un traitement statistique des données sur les divers aspects du rétinoblastome (trop complexe pour être abordé avec des élèves de lycée), que Knudson a proposé une hypothèse sur les mécanismes génétiques à l’origine du rétinoblastome. Cette hypothèse est fournie de façon incomplète dans le document « Origine du rétinoblastome ». Le travail de l’élève consiste alors à voir si les données sur les séquences fournies confirment l’hypothèse de Knudson et amènent à la compléter.
L’hypothèse de knudson repose sur l’idée qu’un seul gène est en jeu et que c’est le même dans les deux cas de rétinoblastome héréditaire et non héréditaire. Surtout, elle suppose que les deux allèles du gène sont mutés dans le cas d’une cellule tumorale. Le génotype d’une cellule atteinte du rétinoblastome suppose donc l’existence de deux mutations, une pour chaque allèle (d’où l’expression « Two hit hypothesis »). Selon cette hypothèse, une cellule ne peut devenir cancéreuse si un seul allèle est muté. Cela signifierait que l’allèle muté est récessif et l’allèle « normal » dominant. C’est contraire à ce que laissait penser l’analyse de l’arbre généalogique de la famille A (mais conforme aux conclusions des expériences de fusion cellulaire).
Durant les années 70 et 80 les recherches se sont poursuivies sur le rétinoblastome jusqu’à aboutir à l’isolement du gène en 1986 et son séquençage en 1987. Après, on a recherché les diverses mutations en cause. Ce sont quelques-unes de ces mutations qui ont été utilisées pour établir les génotypes de quelques individus des arbres généalogiques (Document Origine du rétinoblastome). Pour les individus atteints, on a fourni le génotype des cellules cancéreuses et celui des cellules saines.
Dans le cas du cancer héréditaire, on constate que les cellules cancéreuses ont deux allèles mutés et que les cellules saines de la personne ont un allèle muté et un allèle non muté. En outre un des allèles mutés des cellules cancéreuses est le même que celui trouvé dans les cellules saines. Cela signifie qu’un des allèles mutés a été fourni par un gamète d’un des parents, celui atteint de rétinoblastome, et que le deuxième allèle muté dans les cellules cancéreuses provient d’une mutation somatique apparue dans une cellule rétinienne.
Dans les familles avec rétinoblastome héréditaire, il se peut qu'un individu possède dans toutes ses cellules un allèle muté hérité de l'un des parents et un allèle normal (10 % des cas, non illustré dans l'arbre de la famille A) sans être atteint de rétinoblastome. C'est pourquoi on devrait parler de la transmission d'une prédisposition au rétinoblastome et non de l'hérédité du rétinoblastome. Dans 90% des cas, un tel génotype entraîne l'apparition d'un rétinoblastome. Autrement dit, la probabilité pour qu’un individu ayant dans son génotype un allèle muté et un allèle non muté ne soit pas atteint est très faible (mais non nulle). Si les cellules rétiniennes ont hérité d’un allèle muté, la probabilité pour qu’une mutation intervienne sur l’autre allèle pour une ou quelques-unes des cellules rétiniennes est donc très forte mais elle n'est pas de 100%. Les mécanismes qui font que la possession d'un allèle muté prédispose à une mutation de l'allèle normal sont variés et complexes (mutations ponctuelles comme illustré, délétion, conversion de l'allèle normal en allèle muté, etc.). C’est ce qui explique la possibilité de plusieurs tumeurs dans le cas d’un rétinoblastome héréditaire. Et dans le cas d'un rétinoblastome héréditaire, on a remarqué aussi que la fréquence d’autres cancers, notamment d’ostéosarcomes, est par la suite augmentée.
Pour le cancer sporadique, les individus atteints ont deux allèles mutés dans les cellules cancéreuses, souvent deux allèles différents. En revanche, les autres cellules du corps ont deux allèles non mutés. Cela confirme l’hypothèse de la nécessité de deux allèles mutés du gène RB dans une cellule rétinienne pour qu’un rétinoblastome se développe. Cela est beaucoup moins probable que si la cellule rétinienne a déjà hérité d’un allèle muté et qu’il suffit d’une seule autre mutation somatique pour la rendre cancéreuse.
Dans le cas de la famille C, les parents du premier individu atteint d’un rétinoblastome (III1) possèdent dans toutes leurs cellules deux allèles « normaux » du gène RB ce qui est conforme avec le fait qu’ils n’ont pas été atteints par ce type de cancer durant leur enfance. Cependant leur fils possède dans toutes ses cellules non cancéreuses un allèle muté. Cela signifie que cet allèle muté était présent dès le stade œuf. Il l’a hérité d’un des gamètes qui lui a donné naissance. Il y a donc eu une mutation germinale affectant le gène RB chez un des parents (mutation de novo). A partir de là, la prédisposition au rétinoblastome devient héréditaire dans la famille comme l’illustrent III1 et III3.
On peut en conclusion de ce travail demander aux élèves de schématiser les mécanismes en jeu dans les deux types de rétinoblastome. Les figures ci-dessous illustrent des représentations possibles.
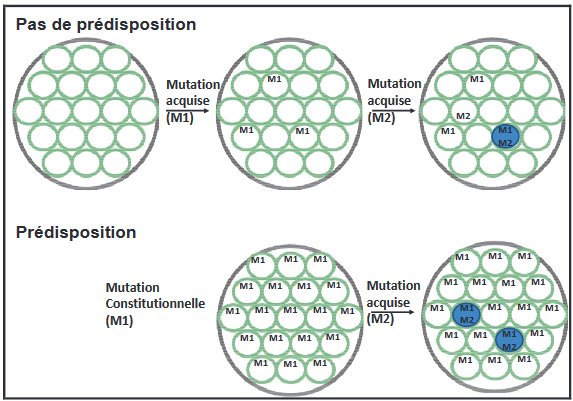
Figure 3 d'après : Etude des relations génotype phénotype dans le rétinoblastome Laurent Castera. Ici sont schématisées des rétines (grands cercles) et des cellules rétiniennes (petits cercles). Les tissus prédisposés portent la première mutation (M1) inactivant le premier allèle dans toutes les cellules. L’apparition d’une deuxième mutation (M2) inactivant le deuxième allèle d’un gène suppresseur de tumeur initie le développement d’un rétinoblastome (cercle bleu).
· La figure ci-dessous résume les mécanismes causes du rétinoblastome héréditaire et non héréditaire.
. 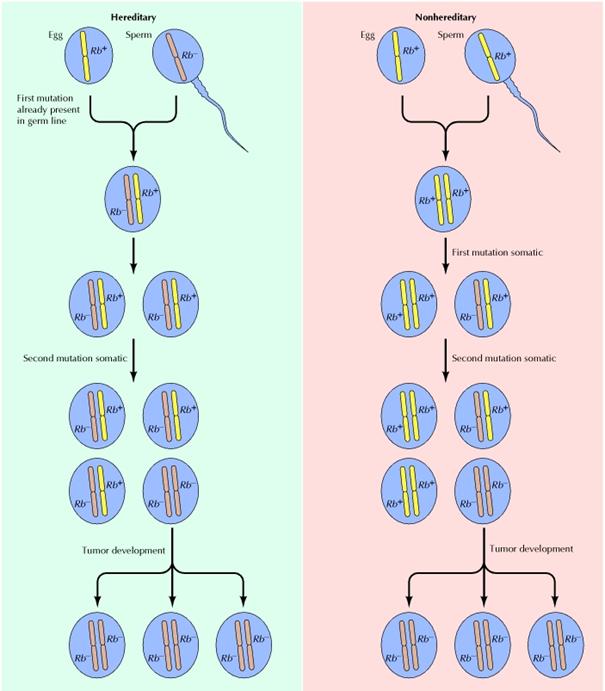
D'après The Cell: A Molecular Approach. Geoffrey M Cooper. Boston University. Sunderland (MA): Sinauer Associates; 2000.
Le gène Rb oncogène ou gène suppresseur de tumeur ?
Les données précédentes indiquent que des mutations du gène Rb peuvent conduire des cellules de la rétine à devenir cancéreuses. Si on a envisagé auparavant la notion d’oncogène, on a tendance à considérer que les allèles mutés de RB sont des oncogènes et l’allèle non muté un proto-oncogène.
Pour tester cette idée, on peut rechercher les caractéristiques des protéines mutées du gène RB en se rappelant que la protéine d’un oncogène présente un gain de fonction par rapport à celle d’un proto-oncogène.
La traduction de l’allèle normal du gène Rb grâce au logiciel Anagène indique que cet allèle code pour une protéine de 928 acides aminés. La traduction des allèles mutés présents dans les familles A, B et C montre que tous ces allèles codent pour une protéine nettement plus courte que la protéine codée par l’allèle normal.

Cela résulte du fait que les mutations sont des mutations non-sens, ou des délétions, insertions de 1 à quelques nucléotides entraînant l’apparition d’un codon stop anticipé. Ces protéines fortement tronquées sont non fonctionnelles. Les mutations de Rb entraînent une perte de fonction du gène. C’est le contraire d’une protéine oncogénique qui est davantage active que la protéine pro-oncogénique normale. Donc le gène RB, bien qu’impliqué dans le rétinoblastome, n’est pas un oncogène ; cela est confirmé par le fait que l’allèle muté RB est récessif par rapport à l’allèle normal alors que c’est l’inverse dans le cas d’un oncogène.
La perte de fonction du gène RB (deux allèles mutés) entraîne dans les cellules rétiniennes l’apparition de tumeur. Cela indique que la fonction normale du gène est de s’opposer à la cancérisation des cellules. C’est ce qui justifie le qualificatif de gène suppresseur de tumeur. Le gène RB, premier gène de ce type isolé et séquencé, en est le prototype.
Une expérience de transfection du gène RB normal dans des cellules tumorales a confirmé que la protéine normale s’oppose à la genèse de tumeur puisque les souris ayant reçu des cellules tumorales avec le gène RB transfecté n’ont pas développé de tumeurs, contrairement à celles chez lesquelles on a injecté des cellules tumorales non modifiées.
Les chercheurs ont réussi à caractériser l’action d’une protéine RB normale. Très schématiquement, la protéine existe sous deux états, l’un déphosphorylé, l’autre phosphorylé. A l’état déphosphorylé, elle s’oppose au passage de la phase G1 à la phase S du cycle cellulaire et donc à la prolifération cellulaire. A l’état phosphorylé, elle ne bloque plus le passage G1-S et donc le déroulement de la mitose. D’autres fonctions de la protéine RB déphosphorylée sont d’intervenir dans la différenciation cellulaire et de stimuler l’apoptose de cellules chez lesquelles l’ADN est endommagé.