
Thérapies cellulaires et géniques : un espoir pour certaines maladies du cerveau
Association Huntington - France
I Aspects cliniques de la maladie de Huntington ou MH (ou Huntington desease, HD)
C'est une maladie génétique
Cette maladie est monogénique, autosomique. Le gène impliqué, nommé IT15, est porté par le bras court du chromosome 4 et est connu depuis une dizaine d'années.
Ce gène code pour une protéine, la huntingtine, dont le rôle est encore à l'étude. La présence d'une protéine huntingtine de type anormal entraîne la formation, dans le noyau de certains neurones (striatum mais aussi autres parties du cerveau), d'agrégats qui pourraient (selon certains auteurs seulement) constituer un des facteurs déclenchant la mort de ces neurones par apoptose.
La protéine anormale doit cette caractéristique à la présence, à l'une de ses extrêmités, d'un nombre d'acides aminés Glutamine supérieur à 35. Dès la présence d'une 36ième Glutamine , la maladie se développe.
La mutation est dominante et sa pénétrance est complète. La présence d'un seul allèle muté suffit pour conduire à la maladie.
Cependant, selon l'allèle muté porté par l'individu, la maladie ne s'exprimera pas toujours au même âge. Plus cet allèle porte un grand nombre de fois le triplet CAG (au dessus de 35), déterminant un nombre identique de glutamine au niveau de la protéine, plus l'âge auquel apparaitra la maladie est précoce. Si la protéine comprend plus de 50 glutamines la maladie s'exprimera très tôt (on a trouvé des nourrrissons porteurs de 180 répétitions chez lesquels la maladie s'est exprimée dès 15 mois).
C'est une maladie qui frappe "violemment"
En quelques semaines, voire quelques mois, toutes les fonctions du cerveau sont atteintes et les symptômes suivants apparaissent:
- troubles de la motricité: certains mouvements sont ralentis alors que d'autres, très rapides, apparaissent (il est possible de bloquer ces derniers par l'usages de neuroleptiques)
- dépression qui n'est pas de type "réactive", elle constitue un des symptômes de cette maladie
- modification du comportement: ces personnes deviennent très irritables
- troubles intellectuels: pertes de mémoire, diminution de la possibilité de parler, baisse de la stratégie (les facultés d'adaptation à l'environnement diminuent)
MH et société
Autrefois, il existait des réactions violentes de la part de l'environnement social de ces malades. L'histoire, il y a 300 ans, des "sorcières de Salem" en témoigne. Certaines de ces femmes pendues ou brûlées en Nouvelle Angleterre étaient atteintes de la MH. C'est grâce aux descendants de ces femmes qui ont pu être retrouvés que ce diagnostic a été porté.
La maladie de Huntington est rare, elle représente environ 3 naissances pour 10 000 (comme la Mucoviscidose).
En France , elle concerne 18 000 personnes: 6000 qui ont atteint l'âge du début de maladie et vivent avec (pendant environ 25 ans) et 12 000, chez lesquels l'allèle morbide a été trouvé mais qui n'ont pas encore de symptômes.
Enfin il faut rajouter à ces personnes 15 000 autres qui sont des personnes faisant partie d'une famille où la MH est présente mais qui ne savent pas que leurs deux allèles sont normaux.
En effet, le diagnostic de dépistage de l'allèle morbide (test génétique) existe, mais la majorité des personnes "à risque" n'a pas envie de connaître son "avenir". Souvent ces personnes savent très bien à quoi s'attendre (mort dans la démence, avec une cachexie, dans des asiles) puisqu'ils ont un parent atteint, et préfèrent vivre dans l'ignorance, et donc aussi l'espoir, "les quelques années qui restent"....
En France moins de 10% des personnes à risque sont allées faire le test. En Hollande, où il existe de grandes compagnies pour cela, ce taux n'a jamais dépassé les 25%.
Le docteur Peschanski a souligné qu'en France, malgré cela, le système social n'a pas vraiment de structure d'accueil pour des adultes entre 25 et 60 ans, handicapés lourdement.
II Aspects biologiques de la MH et essais de thérapie en cours
Les neurones du striatum dégénèrent
Le striatum fait partie des ganglions de la base, relais des voies commandant tout l'organisme (dont les rôles sont en particulier d'exercer un contrôle des fonctions motrices et des activités mentales). Des neurones de la zone frontale et préfrontale du cortex font relai dans le striatum avant de commander les muscles du corps, ainsi , tout ce qui est programmé dans le cerveau doit passer par le striatum avant de prendre effet. Il n'y a pas de suppléance possible.
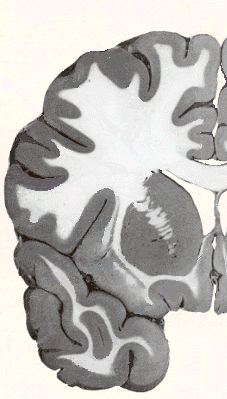
Dès le début des symptômes, les neurones du striatum sont progressivement détruits, plusieurs grades (1 à 4) ont été définis selon la surface de la zone atteinte visible en coupe histologique. Entre le grade 1 et le grade 3 il y a disparition de 80% des neurones du striatum. Certaines personnes jeunes s'étant suicidées dès le début des symptômes, des coupes montrant le grade 0 ont été réalisées et servent de référence.
 |
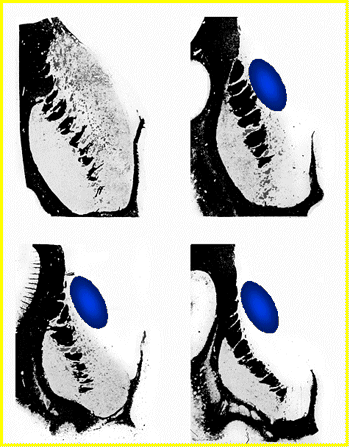 |
| Hémi section coronale (coupe histologique verticale transverse) d'un cerveau humain, montrant la position du striatum au sein de l'hémisphère gauche, tel que présenté en détail ci-contre (partie haute, la tête du noyau caudé et partie basse, le putamen). | Au cours de la progression de la maladie, le striatum, vu ici en coupe coronale (au même niveau que l'image de gauche), perd progressivement de son volume au fur et à mesure de la dégénérescence des neurones qui le composent. Les coupes montrent les grades 0,1, 2 et 3 successifs et, par comparaison, le volume moyen de neurones apporté par une greffe (en bleu). |
Protéger ces neurones de la destruction
Le rôle de la huntingtine reste, jusqu'à maintenant, inconnu (celui que l'on connaît, à savoir la formation d'agrégats dans le noyau des neurones du striatum, n'est pas celui qui provoque la maladie).
Les axes de recherche actuels sont tournés vers l'identifcation de cibles potentielles pour divers médicaments (Récacémide, co-enzyme Q10, Riluzole) parmi les différentes structures cellulaires impliquées dans cette dégénérescence: récepteur membranaire au glutamate (récepteur NMDA), mitochondrie, huntingtine...
L'équipe du Docteur Peschanski, quant à elle, a mis au point une technique assurant la protection des neurones du striatum par le CNTF(Ciliary Neuro-Trophic Factor, substance de la famille des IL6). Il a été montré, sur un modèle animal (poulet) de dégénérescence du striatum (mais qui n'est pas une vraie maladie de Huntington), que le CNTF protège les neurones du ganglion ciliaire de la mort programmée (apoptose). De même, chez le singe, après des injections d'acide1,3 nitropropionique (bloquant des mitochondries) entraînant une destruction spécifique des neurones du striatum, on observe que 80% des neurones sont protégés de ces lésions lorsque le CNTF est présent.
Cependant , le CNTF est une protéine qui ne peut pas passer la barrière hémato-encéphalique et il est donc nécessaire que cette substance soit introduite directement dans le cerveau pour jouer son rôle neuroprotecteur chez les malades. La libération intra-cérébrale du CNTF par des mini pompes pose un double problème, d'une part ces protéines se collent à la paroi des minitubes et les obstruent, d'autre part, à la température du corps, elles sont rapidement dégradées.
De minipompes biologiques ont été mises au point pour palier à ces deux inconvénients. Elles constituent une thérapie génique qui fourni en continu du CNTF, au sein du triatum.
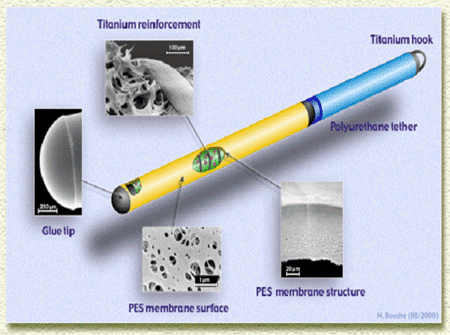
Actuellement la thérapie génique peut se faire soit en utilisant des vecteurs viraux (mais tous les problèmes de sécurité ne sont pas encore résolus), soit des cellules animales que l'on modifie génétiquement. C'est ce type de cellule qui a été cultivé et transformé génétiquement de façon à produire en permanence le CNTF, et à le libérer dans le striatum, une fois encapsulées. Les cellules choisies sont des cellules cancéreuses (ce qui leur confère une relative immortalité), qui ne doivent pas entrer en contact avec les neurones du malade. Elles sont placées dans des capsules (0,6 mm de diamètre et quelques mm de long) dont la paroi poreuse laisse sortir le CNTF qu'elles produisent (ainsi que les nutriments: oxygène et glucose) mais les empêche de passer dans le cerveau du malade. Ces capsules ont été mises au point par Patrick Aebischer, directeur de l'EPFL (Ecole Polytechnique Fédérale de Lausanne, Suisse).
 |
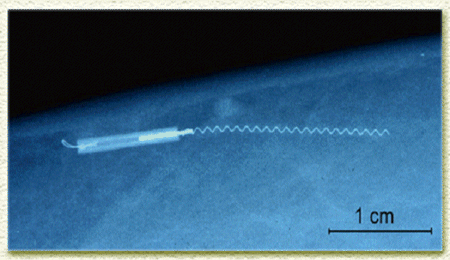 |
| Structure de la capsule permettant un confinement des cellules vivantes tout en laissant passer les molécules solubles. | Radiographie de la capsule (de quelques cm de long), en place dans un tissu vivant. |
Cette technique de macroencapsulation constitue une thérapie cellulaire et génique efficace qui peut fonctionner avec divers types de cellules (de 1989 à 1993 une centaine d'articles ont été publiés par des équipes transformant toutes sortes de cellules par le gène du CNTF), mais ce sont les cellules cancéreuses dont la durée de vie est la plus longue.
Des tests ont été effectué chez les singes. Au moment où les neurones du striatum commencent à mourir (après deux injections d'acide 3 nitropropionique par jour, pendant plusieures mois), les capsules sont mises en place.
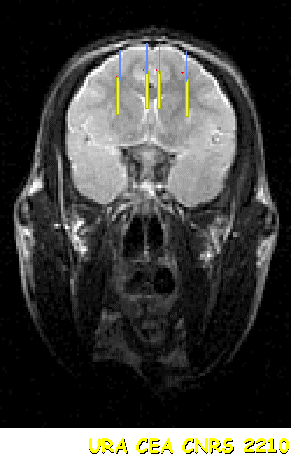 |
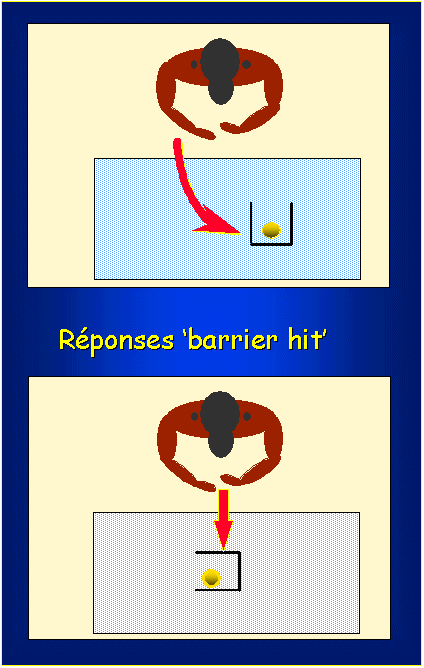 |
| Position des capsules dans le cerveau du singe. IRM sur coupe horizontale. | Test intellectuel d'adaptation stratégique, chez le singe. |
Afin de déterminer si les neurones sont protégés par cet apport de CNTF; des tests moteurs et intellectuels d'adaptation stratégique sont réalisés sur les singes: des sucreries leur sont proposées, placées dans une boîte en plexiglas dont une seule face est absente et qui est positionnée dans n'importe quel sens. Alors qu'un singe normal est capable de s'adapter à une situation nouvelle (changement d'orientation de la boîte), même dans l'obscurité, le singe dont le striatum est atteint présente un comportement "collant" et ne trouvera que par chance l'orifice de la boîte. Ces tests sont quantifiables et permettent de montrer qu'en trois mois de protection par le CNTF des capsules, les singes ont récupéré des performances normales.
Après ces essais chez les singes, le traitement a été appliqué à l'homme. L'implantation d'une capsule a été réalisée, ce qui constitue un test de faisabilité et de sécurité (en réalité, pour le singe, il faut 4 capsules). Ces premiers essais cliniques ont été réalisés avec des cellules (lignée DHK) de hamster nouveau-né, cellules qu'il faut changer tous les 4 mois. Au bout de deux ans, sur les 24 capsules mises en place seulement 11 étaient encore efficaces et c'est pourquoi d'autres cellules que la lignée DHK seront utilisées. Des tests physiologiques ont montré que les malades traités ont présenté des résultats normaux pendant la première année. Cependant lors de l'extraction des capsules (dont ils savaient pourtant qu'elles ne les guériraient pas), plusieurs d'entre eux, considérant alors que tout espoir était perdu, ont montré une dépression réactionnelle.
Remplacer une fraction des neurones atteints
La greffe neuronale de substitution est une autre voie, qui cependant, ne peut constituer qu'un complément thérapeutique. De nombreux travaux avaient montré la faisabilité de la greffe de neurones sur l'animal. Des premières implantations de neurones avaient été faites sur l'homme en 1996.
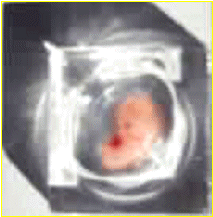 |
 |
| Foetus humain (entre 7 et 14 semaine) fournissant les neurones à greffer. | Mise en place de façon stéréotaxique du greffon d'origine foetale. |
Il n'est pas possible de greffer des neurones adultes car ces derniers ne supportent pas le manque d'oxygène incontournable durant leur transplantation. Il faut se procurer de jeunes neurones humains, en cours de développement, dans des foetus d'âge compris entre 5 et 20 semaines et en particulier entre 7 et 14 semaines pour ceux impliqués dans la Chorée de Huntington. Les cerveaux des foetus extraits au moment d'une interruption volontaire de grossesse sont donc disséqués et leur éminence ganglionnaire coupée en petits fragments qui sont ensuite fournis aux neurochirurgiens. La technique consiste alors à aspirer ces fragments dans une seringue et les injecter au bon endroit (le repérage se fait par image RMN) dans le cerveau du patient. Ces neurones injectés représentent de 5 à 10% des neurones du striatum du malade.
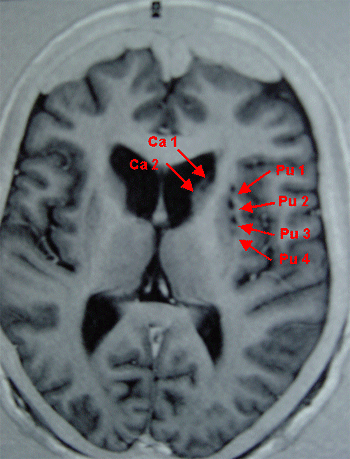 |
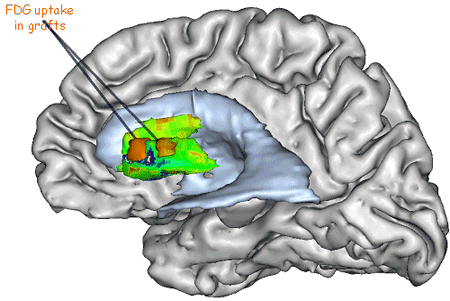 |
Image RMN d'une coupe horizontale de cerveau humain localisant les greffons (deux dans le noyau caudé: Ca1 et Ca2, et 4 dans le putamen: Pu1, Pu2, Pu3 et Pu4) |
L'échelle ci-dessus indique la consommation de FDG (fluoro-désoxy-glucose) visible sur cette image TEP, cette consommation est importante au niveau de deux greffons du striatum. |
Il est possible de vérifier le fonctionnement de ce greffon par l'imagerie TEP: alors que le greffon apparaît en rouge orangé (forte consommation de glucose), le reste du striatum apparaît en vert (faible consommation). Il faut aussi vérifier la bonne connexion de ces neurones avec ceux provenant du cortex, ce sont les tests moteurs qui le révèlent ainsi que les analyses histologiques post-mortem.
Actuellement le docteur Peschanski a greffé 5 malades, ce qui constitue déjà une preuve de concept. Ces malades ont été suivis par des tests de motricité (Bachoud-Lévi et al, 2000) qui mesurent à la fois le nombre de mouvement et leur vitesse. Lors de la dégénérescence liée à la maladie des mouvements rapides apparaissent et certains mouvements volontaires se ralentissent. Les greffes de neurones dans le striatum sont faites sur un côté une première fois, et sur l'autre côté l'année suivante. Mais dès la première greffe on a observé, chez trois malades sur 5, une stagnation des performances motrices, à la place de la baisse attendue en liaison avec la progression de la maladie (l'amélioration est nette pour certains types de problèmes moteurs). Par contre l'effet sur les performances intellectuelles est beaucoup plus difficile à mettre en évidence malgré toutes les précautions d'objectivité prises en amont du test (test MATTIS, test MDRS pour les performances cognitives globales, test TMT-A pour l'attention, ou mesure du potentiel évoqué après stimulation d'un nerf périphérique).
Le passage aux grands nombres
La preuve de concept ayant été apportée, il est maintenant possible de passer à l'échelle d'un plus grand nombre de patients afin d'obtenir une analyse statistique des résultats obtenus. Ceci implique de traiter et de suivre des centaines de malades (ou des dizaines s'ils sont bien encadrés) et de fomer les médecins qui devront, vu le nombre de malades, opérer à la place des chercheurs.
Les médecins formés appartiennent à plusieurs disciplines:
- neurologues (pour les tests)
- neurochirurgiens (pour les greffes)
- obstétriciens (qui récupèrent les foetus)
- des biologistes (pour continuer les recherches)
L'essai en cours, prévu sur 4 ans, doit concerner 70 malades (10 par CHU) francophones (France, Belgique...) et peut-être ira-ton jusqu'à 100 avec des greffes réalisées dans d'autres pays. Un premier groupe de 35 malades doit être greffé immédiatement, puis un autre groupe de 35 sera greffé ensuite. Ces deux groupes seront suivis ensemble pendant 18 mois.
Tout n'est pas aussi simple....
Dans l'étude de concept précedente, deux malades n'ont pas présenté d'amélioration. Chez l'un d'entre eux la greffe n'a jamais "pris", et chez l'autre, après une phase d'amélioration de 5 mois suite à la première greffe, en quelques jours après la deuxième greffe, toute la récupération a été perdue. Il s'était formé un kyste microhémorragique à la place de la greffe (les cellules greffées "pompant" beaucoup de substances nutritives du cerveau ont été "isolées").
Un autre problème est la mort des neurones issus du cortex qui n'arrivent pas à établir de connexion avec les neurones cibles du striatum (ici les neurones greffés).
La question se pose également de savoir si la récupération est suffisamment importante et la rémission suffisamment durable pour justifier le poids de ces greffes. Une récupération uniquement de mouvements oculaires est un faible gain par rapport à la lourdeur du traitement et les problèmes éthiques qu'il pose (utilisation d'embryons humains).
L'association d'une neuroprotection par le CNTF, qui maintient les performances du patient sur un assez long terme, avec une greffe, qui assure une phase d'amélioration des performances motrices sur une durée plus courte, permettrait d'additionner les deux effets bénéfiques et complémentaires.
III Prévention et problèmes éthiques
Il serait possible de vaincre cette maladie en deux générations car il ya fort peu de néomutation. Cette mutation est apparue il y a plusieurs milliers d'années, en Europe de l'Ouest (pendant la période de l'Empire romain). Elle est caractérisée par un effet fondateur fort, en effet on ne la retrouve pratiquement pas en Asie.
Le gène étant bien connu il existe un test génétique permettant de savoir si un individu est porteur de l'allèle malade et déclenchera donc la maladie (rappel: l'allèle étant dominant, il suffit qu'un seul des deux allèles de ce gène soit muté pour que la maladie survienne).
Il est possible d'éviter que des personnes à risque (présence d'un malade dans la famille) transmettent la maladie. Il leur est proposé d'avoir recours à la fécondation in vitro pour concevoir leur enfant. Les embryons créés sont testés pour le gène de la maladie de Huntington (on réalise le test génétique sur une de leurs cellules, c'est un diagnostic pré-implantatoire ou DPI) et on ne transfère dans l'utérus de la femme que des embryons dépourvus de l'allèle morbide. Cette technique évite d'avoir à interrompre une grossesse en cours, ce qui est le cas, lorsque le test génétique est réalisé plus tardivement (diagnostic pré-natal ou DPN) et s'avère positif. Par contre, vu que le gène est dominant, le fait de savoir qu'un de leurs embryons est atteint informe ces personnes sur leur possession de l'allèle morbide, ce que certains n'ont pas envie de savoir.
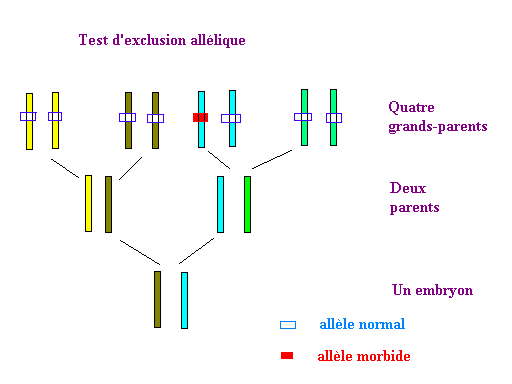
Il est possible d'assurer ces personnes qu'elles auront des enfants normaux, sans avoir connaissance de leur génome, en réalisant une "exclusion allélique": on cherche chez l'embryon la présence d'un chromosome 4 du grand-parent malade (on se fonde sur l'analyse de certains marqueurs très spécifiques), il suffit d'analyser les deux chromosomes 4 du grands parents malades et ceux de l'embryon. Si la réponse est positive, l'embryon a encore un risque sur deux (comme dans le cas du schéma ci-dessous) d'avoir reçu de ce grand parent le chromosome porteur de l'allèle morbide. Mais par précaution, dans ce cas, l'embryon n'est pas implanté dans l'utérus de la mère.
 |
Dès qu'il existera une thérapie efficace pour cette maladie, les individus à risque seront volontaires pour se faire dépister, les gens auront un bénéfice direct à connaître leur risque et l'on sera dans une situation de médicalisation "normale". Une évolution des comportements similaire a été observée dans le cas du SIDA, avec l'apparition de la tri-thérapie, alors qu'au début de cette épidémie, les gens préféraient vivre dans l'ignorance.
Les questions posées par l'assistance
Y a t-il des rejets des greffes de neurones?
Chez l'animal, il n'y a pas de rejet des allogreffes non syngéniques (greffon issu de donneur appartenant à la même espèce mais pas la même lignée). Chez l'homme, des précautions sont prises de façon à limiter ces rejets: on administre des immuno-suppresseurs aux malades greffés, pendant un temps assez court ( 6 mois) correspondant à une réponse immunitaire possible, déclenchée par la brêche vasculaire.
Cependant ce traitement est beaucoup plus léger que ce qui est pratiqué dans les cas de greffes d'autres organes, comme un rein. Le cerveau est en effet un organe qui possède fort peu de défenses immunitaires.
De plus la nécessité de ce traitement ne semble pas prouvée, les malades de Huntington ont un comportement assez "fantaisiste" qui fait qu'ils oublient souvent de prendre leur médicaments (contrairement aux Parkinsonniens qui sont "hyper-méthodiques") et cela ne semble pas avoir de réelles conséquences.
Les greffes sont-elles possibles pour les malades atteints de neurodégénérescence diffuses?
Les cellules prélevées sur les foetus ne peuvent pas circuler dans tout le cerveau, c'est pourtant ce qu'il faudrait pour pouvoir soigner une neurodégénérescence diffuse comme une démence ou une maladie d'Alzheimer. Seules les cellules souches embryonnaires (moins différenciées) peuvent circuler dans le parenchyme cérébral adulte . Cette propriété a été montré chez la souris. Le problème est que ces cellules ont la capacité de donner n'importe quel tissu et même de donner des tumeurs. En général les cellules souches doivent être différenciées en culture avant d'être greffées, mais l'orientation de la différenciation d'une cellule souche en culture (par des substances chimiques) est encore en phase d'étude.
La neuroprotection est-elle envisageable dans le cas du Parkinson?
Concernant la maladie de Parkinson, les tests de neuroprotection qui vont commencer en Suisse utilisent le GNDF, dans le même type de capsules que le CNTF. Cependant, lorsque le Parkinson apparaît, cela correspond en général à une perte déjà importante des neurones du striatum (environ 70 à 80%). En effet, ces neurones n'ayant pas de fonction géographique très précise (leur rôle est de réaliser une "imprégnation locale" de Dopamine plutôt que de se connecter spécifiquement avec d'autres neurones), il peut y avoir compensation par d'autres neurones avant que les symptômes ne soient perceptibles. Le test de neuroprotection ne concernerait donc qu'un petit pourcentage de neurones, seulement 20%....
Prix Littéraires du MEDEC 2003: Grand Prix d'Humanisme médical à
Jean BAREMA
" Le test " (Lattès)
Est-il ou n'est-il pas frappé par une maladie génétique inguérissable ? Tel est le thème de ce passionnant témoignage d'un homme, père de famille, qui attend le résultat d'un test - verdict qui répondra à la question qu'avec angoisse il se pose et qui scellera son avenir.
Association Huntington -France
14 route de Gomez
91440 Bures sur Yvette
http://www.orpha.net/nestasso/HUNTING/