
La diversité génétique du sol
L'étude de la biodiversité microbienne du sol par culture sur boîte de Pétri ne révèle que les espèces cultivables. Or, il existe dans le sol une diversité non cultivable (au moins non cultivable actuellement) que seuls les outils moléculaires permettent de révéler.
Les outils moléculaires
L'étude moléculaire des grands groupes de champignons se fait grâce à des séquences nucléotidiques d'ITS (espaceur interne transcrit, angl. internal transcribed spacer) des unités ribosomiques. les ITS permettent de différencier les espèces : c'est précisément notre objectif, identifier les espèces grâce à la spécificité des séquences nucléotidiques. C'est aussi fondamentalement le principe qui guide la démarche de "barcoding" . Pas trop distincts néanmoins, de sorte que l'alignement des séquences des ITS des différentes espèces reste possible (tout en restant assuré que ce que l'on compare ce sont bien des choses comparables, des séquences homologues suivant les règles de l'analyse de parenté ).
Les trois grands groupes de Champignons (Eumycètes)
|
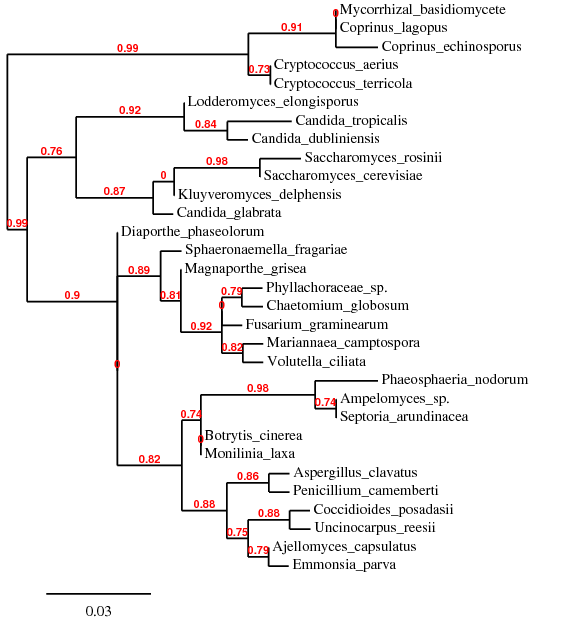
L'alignement des séquences d'ITS de différents champignons permet après construction d'un arbre phylogénétique de repérer 3 grands groupes:
Banque principale de séquences (fichier de séquences au format fasta).
|
 |
|
Traitement hors ligne Il peut être réalisé avec différent logiciel dont Phylogene généralement utilisé dans le secondaire (lien vers téléchargement du logiciel) Rmq. l'analyse des séquences peut être réalisée avec de nombreux outils-logiciels : toutefois nous devons souligner que les résultats des alignements de séquences et les constructions d'arbres phylogénétiques correspondants seront très variables et que seule l'analyse en ligne sur le site MABL (ci-après) donne les résultats que valident les spécialistes. C'est donc cette dernière configuration d'analyse que nous recommandons. |
[adaptation au logiciel Phylogene en cours] |
|
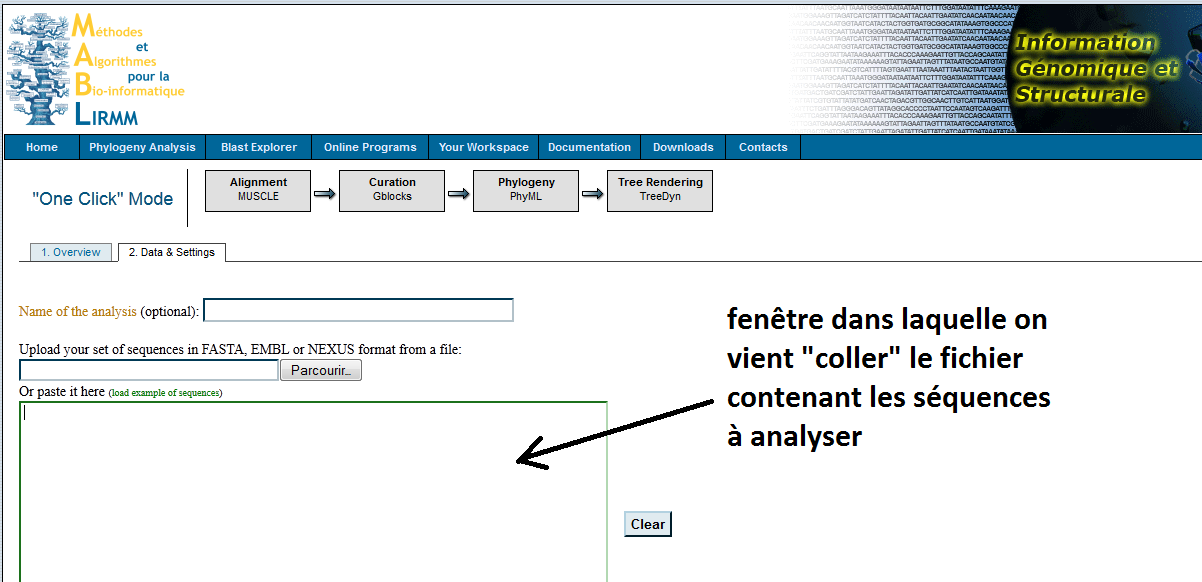
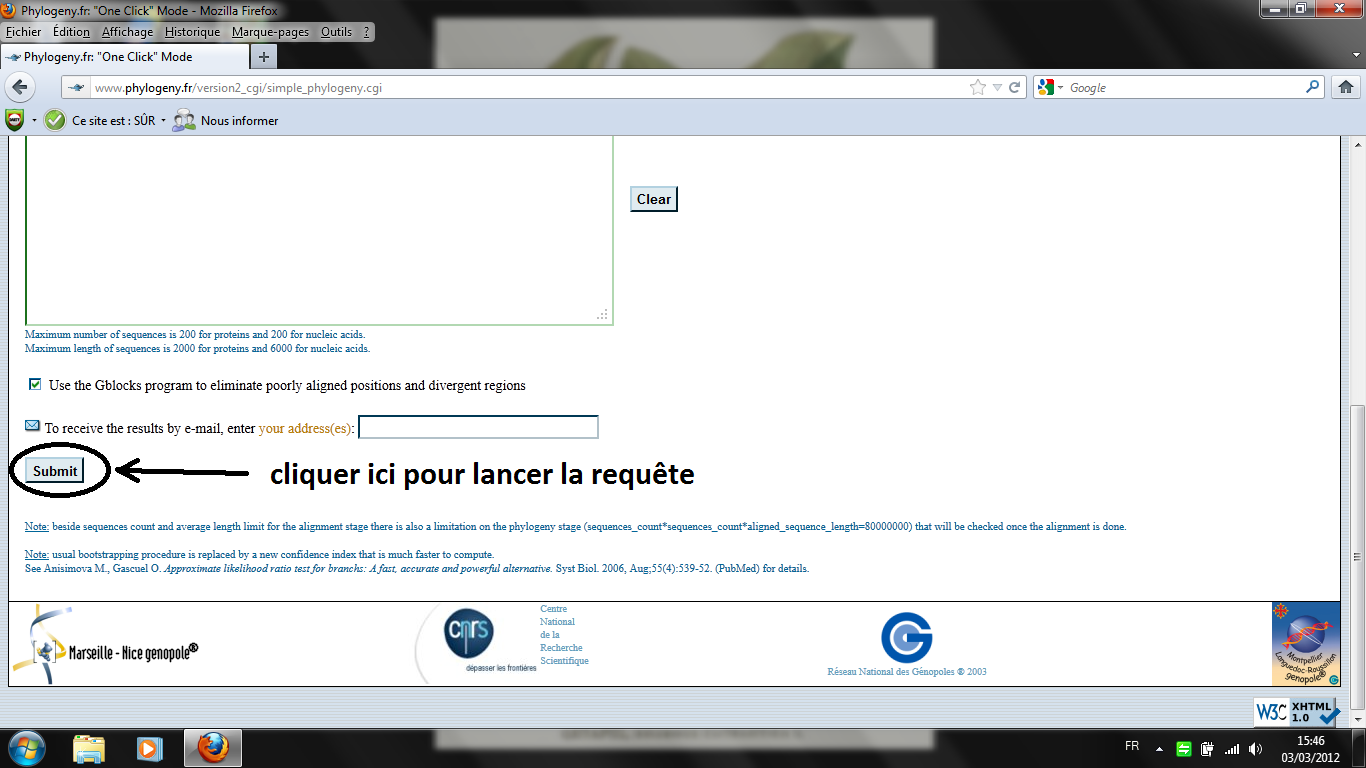
Traitement en ligne Nous proposons de mener ce type d'analyse sur le site de l'Université de Marseille. C'est un excellent outil qui par ses fonctions élaborées permet de "gommer" les nombreux défauts que peuvent présenter les séquences extraites en l'état des banques de données (variations de longueur, de point de départ, de point d'arrivée, nucléotides ambigus, etc.). A noter que cette démarche d'analyse en ligne sur des sites spécialisés et dispersés sur l'ensemble de la planète est de plus en plus fréquente chez les scientifiques. Procédure à suivre : adresse du site ci-contre ; écran d'accueil ; sélection du mode "one click" (situé un peu plus bas sur l'écran d'accueil) ; sélection (copier) du fichier contenant les séquences (la banque principale ci-avant) ; coller les séquences ; soumettre la requête (au pied de la fenêtre où l'on a collé les séquences). Attendre l'affichage de l'arbre. Le temps peut paraître long mais sachez que cet outil est en réalité extraordinairement rapide!!! Les scores (indiqués en rouge, mais il est possible de les supprimer ou de les afficher d'une autre couleur) sont compris entre 0 et 1 (1 pour l'identité) : on considère qu'un score inférieur à 0,7 n'a pas grande signification et ne permet pas de conclure valablement sur une parenté. |
 |
Validation de la banque par des séquences supplémentaires
Afin de vérifier la fiabilité de la banque principale, on se propose "d'injecter" des séquences supplémentaires connues. Le positionnement de ces séquences supplémentaires permettra de vérifier la pertinence de la banque principale. Ceci afin de pouvoir l'utiliser comme outil de détermination de séquences inconnues.
|
L'analyse d'une banque comprenant des espèces nouvelles connues doit permettre de valider l'arbre précédemment construit. Les espèces ajoutées: Les Euascomycètes Sclerotinia minor, Colletotrichum gloeosporioides, Histoplasma capsulatum, Penicillium vulpinum, Sclerotinia sclerotiorum, Fusarium culmorum, Gaeumannomyces graminis, Leptosphaeria sp., Metarhizium anisopliae Les Hemiascomycètes Candida albicans, Saccharomycete sp., Saccharomyces servazzii Les Basidiomycètes Filobasidium capsuligenum
|
Les taxons en vert sont les nouveaux taxons tests |
|
Traitement hors ligne Ouvrir le fichier avec le logiciel Phylogene. Sélectionnez les unes après les autres les séquences nouvelles et afficher l'arbre.
|
[adaptation au logiciel Phylogene en cours] |
|
Traitement en ligne Même démarche que précédemment avec l'option de sélectionner une à une les nouvelles séquences et les ajouter à la banque principale, ou prendre l'ensemble des nouvelles séquences (l'ensemble du fichier) et les ajouter à la banque principale. On pourra comme dans l'image proposée en illustration ci-dessus affecter une couleur particulière aux nouvelles séquences testées. |
|

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Localisation de séquences inconnues dans l'arbre des champignons
Lors de l'étude d'un sol, on extrait des séquences inconnues et le traitement de ces séquences dans la banque principale de séquences permet de déterminer le groupe auquel appartiennent ces séquences inconnues. Nous conservons par la suite le libellé anglais uncultured car c'est sous cette forme que nous trouvons ce type de séquences dans les banques de séquences internationales.
Une banque supplémentaire de séquences d'espèces non cultivées. |
 |
|
Traitement hors ligne
Ouvrir le fichier avec le logiciel Phylogene. Sélectionnez les séquences uncultured et afficher l'arbre.
|
[adaptation au logiciel Phylogene en cours] |
|
Traitement en ligne
Même démarche que précédemment
|
|
Rmq. :
- On retiendra le caractère relatif de ces arbres dont les résultats dépendent de différents choix parfois plus ou moins arbitraires si l'on n'y prend pas suffisamment garde.
- Au-delà on retiendra aussi que ce sont de fabuleux outils qui ont contribué au profond renouvellement et même bouleversement qu'a connu récemment la classification du monde vivant.
- C'est aussi (c'était là notre objectif) ainsi que les scientifiques évaluent la présence d'êtres vivants que personne n'a encore réussi à observer ou cultiver, et qui sont les plus nombreux puisqu'on estime à tout au plus 1% la biodiversité microbienne connue (au sens classique de la description des espèces).
- Ex. de document publié par les scientifiques : il s'agit d'une étude sur les champignons saprophytes et mycorhiziens d'une forêt boréale publiée dans la revue New Phytologist dont les parutions anciennes sont gratuitement accessibles en ligne ; la fig.4 de cet article de Lindahl et coll. montre un arbre phylogénétique où l'on voit la part importante de ce qui doit être majoritairement des "uncultured" représentés par un code et non par une désignation binôminale ; télécharger le fichier .pdf pour obtenir un zoom de l'arbre et visualiser les noms des espèces.