
Thérapie génique par édition du génome
IV. Thérapie génique par édition du génome
A - Données scientifiques et techniques en relation avec la thérapie génique par édition du génome.
1 - Données scientifiques
Les données sur la mutation du gène bêta A à l’origine de la drépanocytose (SCD) ont été précédemment rappelées. Une autre connaissance importante se rapporte à la production de l’hémoglobine foetale qui dépend de l’expression du gène Gamma. Une des propriétés de cette hémoglobine foetale est d’inhiber plus ou moins fortement en fonction de sa concentration la polymérisation de l’hémoglobine drépanocytaire HbS et donc de s’opposer aux symptômes de la drépanocytose. Ainsi, ceux-ci n’apparaissent chez les enfants drépanocytaires que 3 mois environ après la naissance, lorsque le gène gamma cesse de s’exprimer.
![]()
Source :
Transition from fetal to adult hemoglobin
Le mécanisme cause de l’arrêt de l’expression du gène Gamma est complexe ; le gène BCL11A, situé sur le chromosome 2, y joue un rôle important. Il code pour un facteur de transcription qui inhibe l’expression du gène gamma. Ce gène est « silencieux » pendant la vie foetale et commence donc à s’exprimer après la naissance ; son expression dépend d’une séquence activatrice, un « enhancer » spécifique à la lignée érythrocytaire.
Deux stratégies d’édition du génome s’appuient sur ces données. La première vise à réactiver la production d’HbF chez la personne drépanocytaire en réactivant l’expression du gène gamma. Pour cela, elle vise à inactiver l’expression du gène BCL11A et par là à supprimer l’inhibition qu’il exerce sur celle du gène gamma.
La deuxième stratégie plus directe vise à corriger dans les cellules souches hématopoïétiques, la mutation beta S, donc à rétablir des allèles fonctionnels beta A.
2 - Données techniques
Le système CRISPR-Cas9 est le plus utilisé dans la thérapie génique d’édition du génome.
La description et les fonctions des composants de ce système sont décrites dans le dossier sur la maladie de Steinert : « Principe d’une thérapie génique utilisant le système CRISPR-Cas9 ». La figure ci-dessous schématise ce système en action, comment il contribue à modifier une séquence nucléotidique.
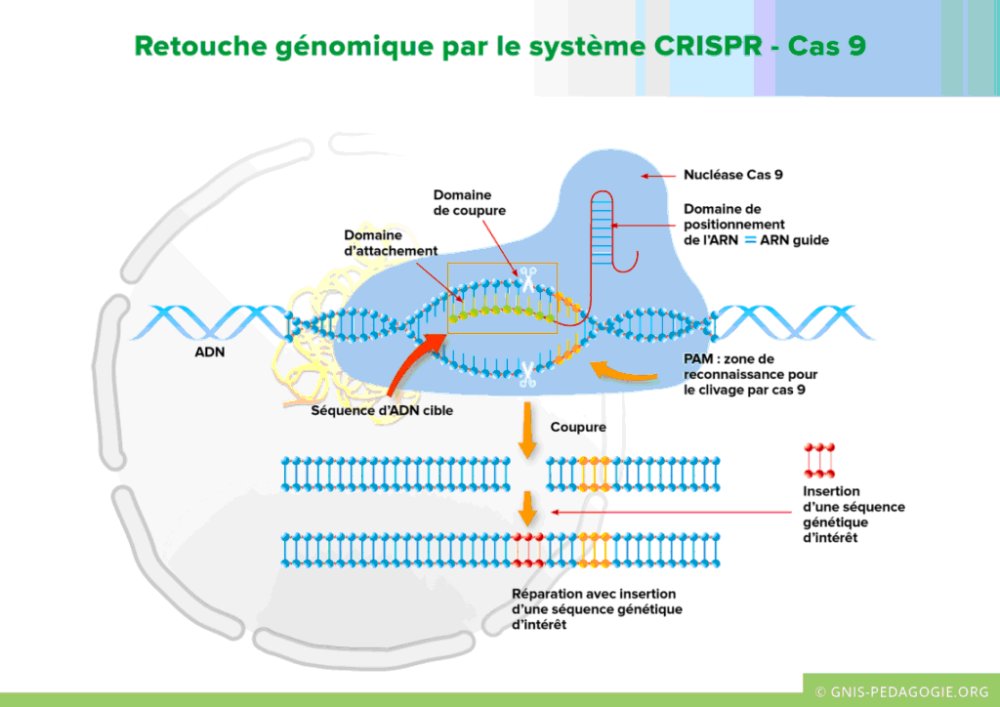
Source :
La figure montre l’action du système Crisper-Cas9 dans le noyau d’une cellule sur une séquence nucléotidique : il provoque une cassure double brin en un endroit précis de la séquence. Suite à cette coupure, on peut insérer une séquence nucléotidique d’intérêt et ainsi modifier la séquence et sa fonction. La cellule dispose d’un autre mécanisme de réparation de la cassure, qui introduit des insertions et délétions autour du site de coupure, ce qui a pour conséquence d’inactiver la séquence (gène ou séquence régulatrice).
La coupure de la séquence cible est réalisée par le complexe formé par l’enzyme Cas et un ARN guide. Une séquence de 3 nucléotides - appelée PAM - de l’ADN à couper par le complexe ARN guide Cas9, initie la séparation des deux brins de la région cible à modifier. L’ARN guide débute par une séquence de 20 nucléotides qui est complémentaire de celle de la séquence cible. L’enzyme Cas9 - qui est une nucléase- guidée par l’ARN, peut alors couper la séquence cible à un endroit précis, situé 3 nucléotide en amont de PAM.
B - Thérapie génique de la drépanocytose par réactivation de la production d’hémoglobine foetale HbF.
En janvier 2021, dans un article du « New england journal of medicine : CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. Haydar Frangoul et al., N Engl J Med 2021; 384:252-260 » une équipe de médecins dirigée par Frangoul fait part des résultats positifs obtenus chez un patient drépanocytaire, avec une thérapie par édition génomique. Cette thérapie est basée sur la connaissance de l’inhibition exercée par le gène BCL11A sur l’expression du gène codant pou la chaîne gamma.
1 - La patiente et le protocole global de la thérapie
La patiente est une femme de 33 ans qui, durant les deux années précédant la thérapie, a eu en moyenne 7 crises vaso-occlusives très douloureuses. Elle a reçu 5 transfusions de globules rouges par année durant cette période de deux ans (voir figure F sur les résultats plus loin). Elle soufre donc d’une drépanocytose sévère.
La figure ci-dessous récapitule la succession des étapes de la thérapie de la patiente.
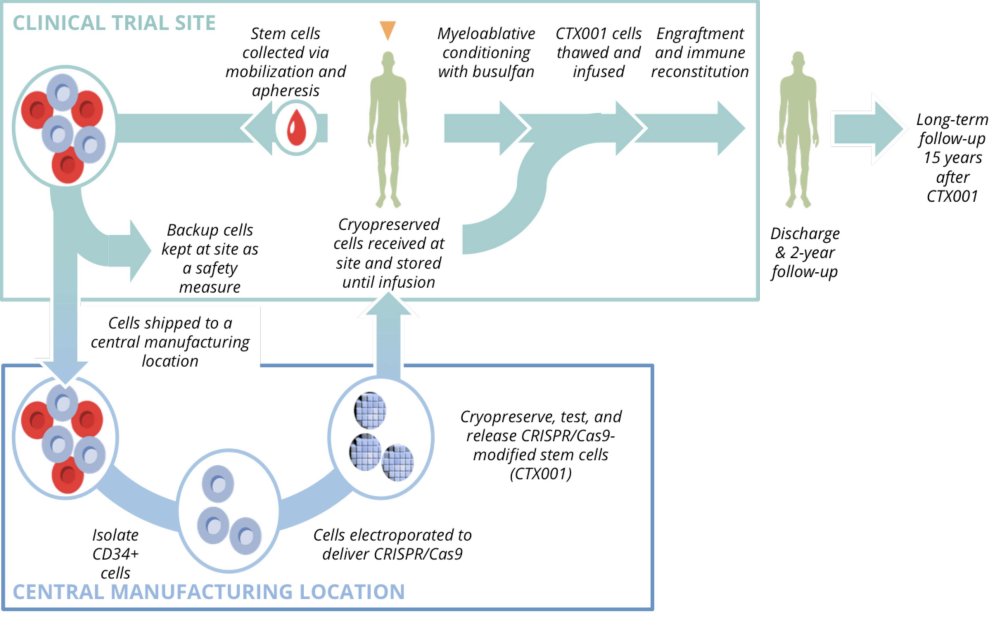
Source : « New england journal of medicine : CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. Haydar Frangoul et al., N Engl J Med 2021; 384:252-260 »
- Recueil des cellules souches hématopoiétiques de la patiente à l’hôpital.
- Stockage d’une partie des cellules souches à l’hôpital par mesure de sécurité.
- Transfert des cellules souches au laboratoire chargé de les modifier génétiquement.
- Electroporation des cellules souches permettant d’introduire les composants de CRISPR-Cas9 dans ces cellules.
- Récupération des cellules modifiées sous forme d’un produit appelé CTX001.
- Transfert de CTX001 à l’hôpital.
- Conditionnement myéloablatif de la patiente par une chimiothérapie (Busulfan), capable de détruire les cellules souches de génotype bêta S//bêta S de la moelle osseuse, et ainsi permettre que leurs places soient prises par les cellules génétiquement corrigées.
- Injection des cellules génétiquement modifiées.
- Engraftment, processus par lequel les cellules souches hématopoiétiques génétiquement modifiées gagnent la moelle osseuse où elles seront à l’origine des futurs globules rouges (multiplication et différenciation).
- Suivi à court, moyen et longt terme de la thérapie génique.
- CTXOOI produit injecté à la patiente, désigne les cellules autologues CD34+ dont le génome a été modifié par CRISPR-Cas9 au niveau de l’enhancer spécifique du gène BCL11A.
2 - Modifications génétiques des CSH de la patiente
La figure ci-dessous indique que cette modification génétique a été obtenue eu utilisant le système CRISPR-Cas9.
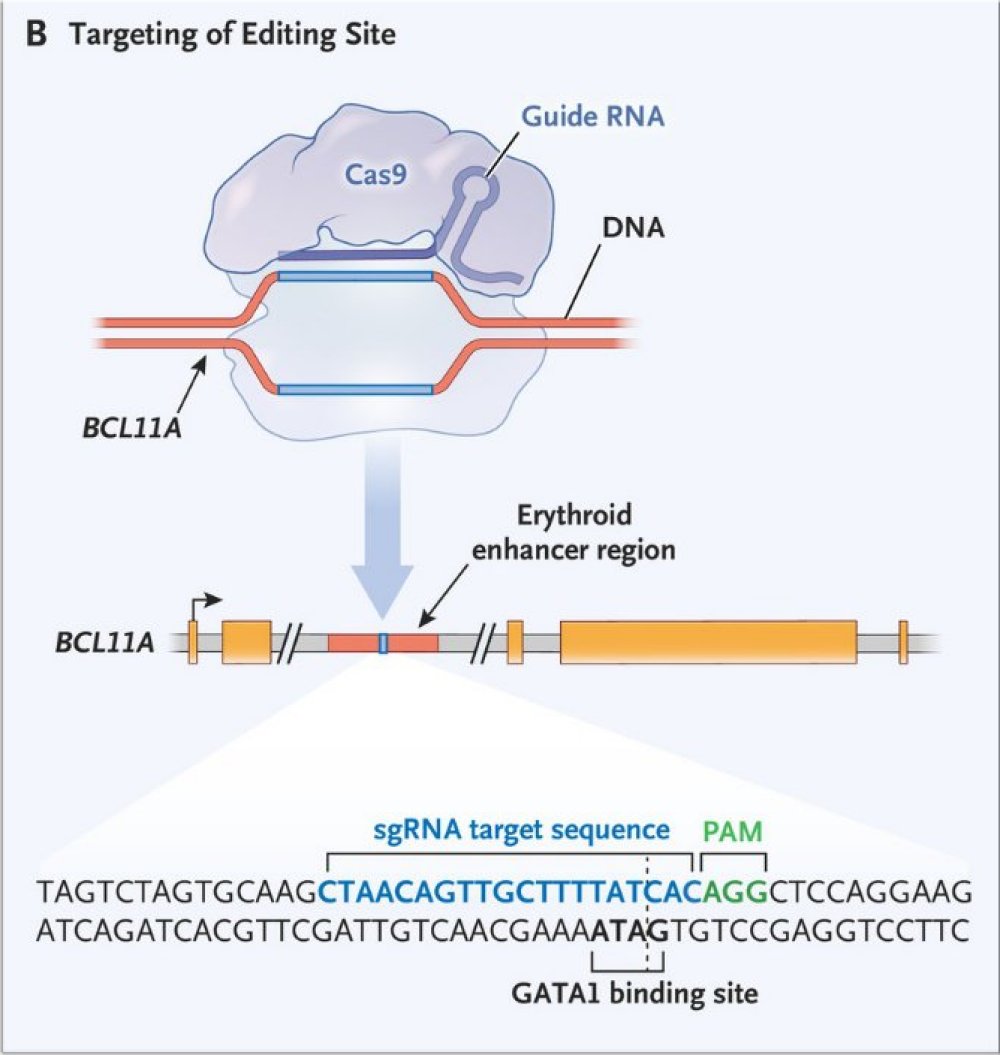
Source : « New england journal of medicine : CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. Haydar Frangoul et al., N Engl J Med 2021; 384:252-260 »
Il faut d’abord introduire les constituants du système d’édition dans les CSH. L’équipe de médecins n’a pas utilisé un vecteur mais la technique d’électroporation. C’est une technique où on soumet les cellules à un champ électrique. Cela a pour effet de créer des pores temporaires dans les membranes cellulaires à travers lesquels, des molécules comme les acides nucléiques, ADN et ARN peuvent pénétrer dans la cellule.
La figure montre que CRISPR-Cas9 va agir sur le gène BCL11A et plus précisément sur un enhancer spécifique de ce gène dans le génome des CSH.
Un enhancer, qu’on peut traduire par amplificateur, est une séquence non codante stimulatrice de l’expression d’un gène, de sa transcription. L’enhancer du BCL11A des CSH est situé dans le deuxième intron de ce gène. D’une manière générale , les enhancers des gènes qui sont des séquences régulatrices sont situés à des distances variables de la séquence du gène, en amont, en aval ou même dans un intron d’un gène comme c’est le cas pour BCL11A illustré dans la figure.
En bas de la figure B, la séquence d’ADN double brin de l’enhancer est fournie. On indique aussi la position de l’ARN guide d’une vingtaine de nucléotides qui est complémentaire d’un brin de l’ADN cible de l’enhancer. Guidé par cet ARN, la nucléase Cas9 va réaliser des cassures double brin de l’ADN de l’enhancer.
La cellule dispose de systèmes de réparation des cassures de l’ADN. Dans le cas de l’enhancer, le système de réparation, appelé NHEJ, assure la jonction entre les deux extrémités non homologues. Ce faisant, il introduit des courtes délétions ou insertions modifiant la séquence initiale de l’enhancer. Celles-ci font que la fonction de l’enhancer est abolie ou fortement réduite. Cela supprime ou diminue considérablement la transcription du gène BCL11A. En conséquence, l’expression du gène gamma de l’hémoglobine est moins réprimée, ce qui entraîne une augmentation de l’hémoglobine foetale.
3 - Evolution de paramètres biologiques et cliniques suite à la thérapie par édition du génome
Dans leur article, les médecins de l’équipe de Frangoul font part de l’évolution des paramètres suivants chez la patiente ; Taux d’hémoglobine totale ; fraction de chaque type d’hémoglobine (HBA ; HbF ; HbS ; HbA2 ) par rapport à l’hémoglobine totale ; pourcentage de cellules F, hématies possédant de l’hémoglobine F. En outre, ils renseignent sur les transfusions de globules rouges reçues ainsi que sur les crises vaso-occlusives ayant affecté la patiente avant la thérapie et durant les 16 mois qui ont suivi. Les graphiques ci-dessous illustrent l’évolution de ces paramètres.
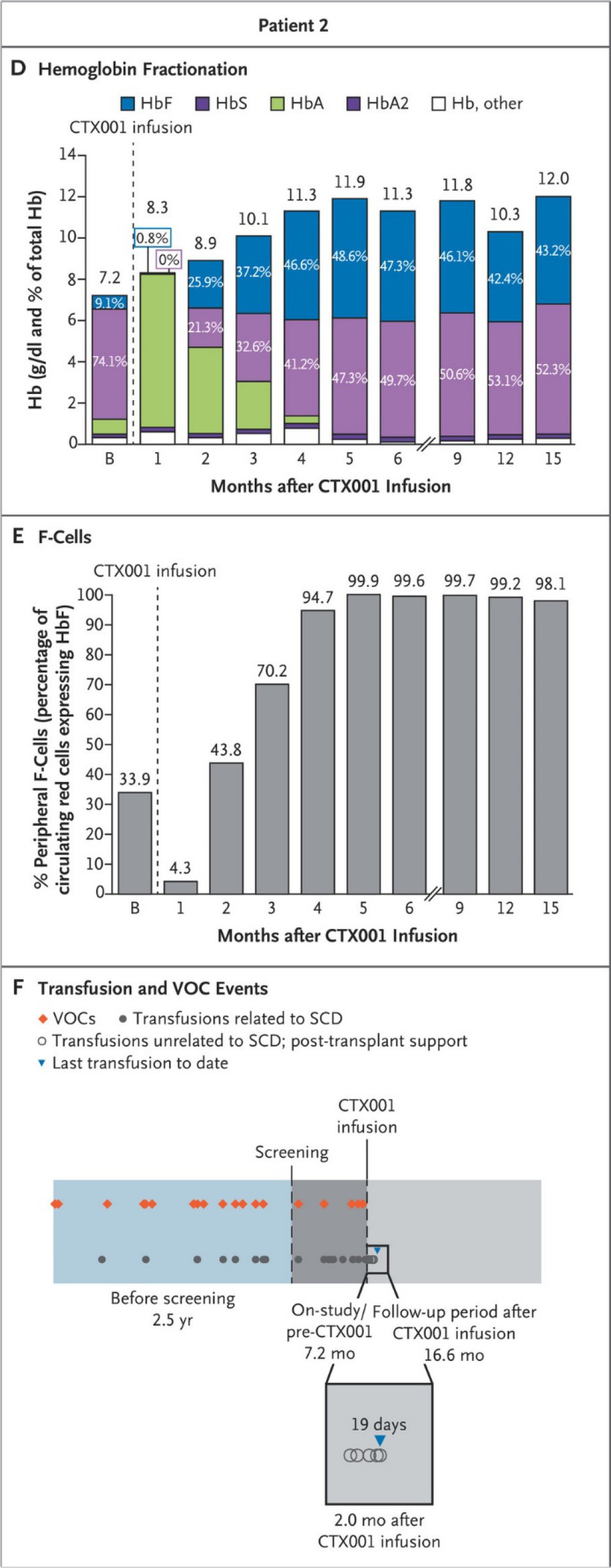
Source : « New england journal of medicine : CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. Haydar Frangoul et al., N Engl J Med 2021; 384:252-260 »
La figure D indique l’évolution de la quantité totale d’hémoglobine du sang (au dessus de chaque rectangle) et le pourcentage de chaque type d’hémoglobine par rapport à l’hémoglobine totale.
La figure E indique le pourcentage d’hématies F possédant de l’hémoglobine foetale.
La figure F a trait à des données cliniques.
Les crises vaso-occlusives sont indiquées en rouge ; les transfusions sont en bleu ; la dernière transfusion a été administrée 19 jours après l’injection de CTX001.
L’anémie est une caractéristique de la drépanocytose due à une destruction précoce des hématies en faucille. Le taux d’hémoglobine totale de 7,2 g/dl est nettement inférieur à la fourchette de valeurs considérées comme normales. Cette hémoglobine est essentiellement de l’hémoglobine S (74,1%). Le pourcentage d’HbF est de 9,1%. Il est supérieur à celui trouvé chez les personnes non drépanocytaires (1%). C’est une caractéristique commune aux drépanocytaires mais cela est insuffisant pour atténuer les symptômes de la maladie.
Au moment de l’injection de CTX001, la quantité totale d’hémoglobine a augmenté puisqu’elle est de 8,3g/dl et c’est essentiellement de l’HbA. Celle-ci n’a pas été produite par la patiente qui a le génotype bêta S//bêta S. Elle provient des transfusions reçues pendant la phase de conditionnement précédant l’injection de CTX001. A partir du 4ème mois, la quantité totale d’hémoglobine varie entre 11 et 12 g//dl, valeurs considérées dans l’échelle basse des valeurs normales. A 15 mois, le pourcentage de HbS est légèrement supérieur à celui de HbF mais la valeur de ce dernier traduit par rapport à la situation initiale une augmentation importante de l’hémoglobine foetale ce qui était le premier objectif de la thérapie.
Les données sur le pourcentage d’érythrocytes (F-Cells) possédant de l’HbF indiquent qu’à partir de 4 mois après la thérapie le pourcentage d’hématies produisant de l’HbF est près de 100% alors qu’il n’était que de 33% avant la thérapie. Cela témoigne du fait que presque toutes les hématies produites dérivent de cellules CSH dont le génome a été modifié. Toutes les cellules de CTX001 injectées n’avaient pas un génome modifié mais celles qui l’avaient et les cellules qui en dérivent ont eu un avantage sélectif de sorte que toute la population d’hématies de la patiente a produit de l’HbF.
Les données cliniques indiquent que la patiente n’a plus reçu de transfusions sanguines à partir de 19 jours après la thérapie et qu’elle n’a souffert d’aucune crise vaso-occlusive. Les données cliniques confirment donc l’efficacité de cette thérapie génique axée sur l’augmentation de la production d’hémoglobine HbF chez la patiente. Toutefois les médecins auteurs de cette thérapie insistent sur le fait qu’il faut la réaliser sur une cohorte de patients et voir si ses effets se maintiennent à long terme et sans apparition de complications secondaires plus ou moins graves.