
Données ayant conduit à concevoir une thérapie génique innovante
II - Données ayant conduit à concevoir une thérapie génique innovante
A - Des précisions sur la structure de la dystrophine
L’objectif de la thérapie génique de la myopathie de Duchenne est d’introduire dans les fibres musculaires un gène DMD qui code pour une dystrophine fonctionnelle. Les propriétés essentielles de la protéine ont été illustrées par ailleurs. La figure « les domaines de la dystrophine » fournit des informations supplémentaires pour comprendre les caractéristiques du gène thérapeutique.
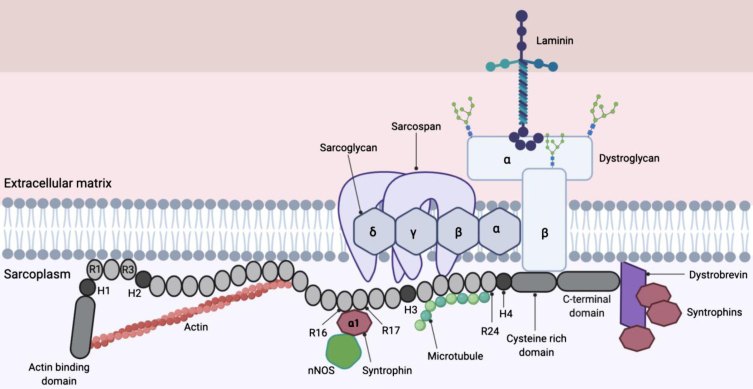
Source: Nertiyan Elangkovan and George Dickson. Gene Therapy for Duchenne Muscular Dystrophy. J Neuromuscul Dis. 2021.
Les domaines de la dystrophine
La dystrophine est une protéine de 3685 acides aminés qui se localise juste sous la membrane (sarcolemme) de la fibre musculaire. Elle assure une liaison solide (continuité mécanique) entre la matrice extracellulaire et le cytosquelette intracellulaire (actine) par l’intermédiaire d’un complexe de protéines membranaires (Dystroglycanes, sarcoglycanes, sarcospanes) et cytoplasmiques (Dystrobrevines et Syntrophines).
On distingue 4 domaines structuraux distincts : le domaine N-terminal de liaison à l’actine (acting binding domain, ABD) ; le domaine central « rod » composé de 24 unités répétées (R1 à R 24), le domaine riche en cystéines (acide aminé) qui assure le lien avec le complexe de protéines membranaires ; le domaine terminal qui se lie aux protéines cytoplasmiques du complexe. Quatre régions charnières flexibles (H1-H4) interrompent le domaine « rod ».
Le schéma sur la structure du gène permet de situer les domaines par rapport à la séquence des exons.

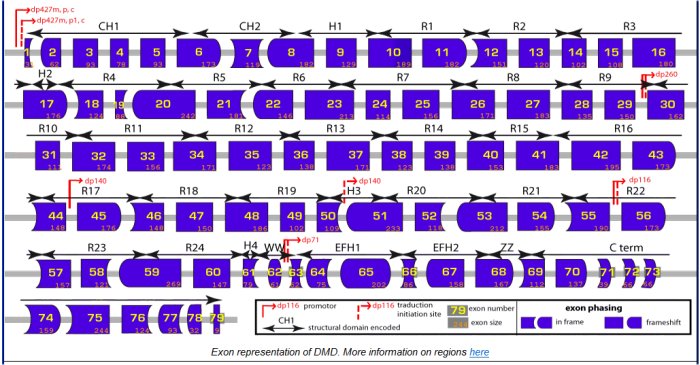
Source : e-dystrophin database. The gene.
B - Le problème de la thérapie génique lié aux caractéristiques du gène DMD
Le gène DMD avec ses 79 exons et 78 introns est le plus grand gène du génome humain. Sa séquence complète fait plus de 2 millions de paires de bases et sa séquence codante 11200 paires de bases. Même l’ADN de la séquence codante ne peut être contenu en entier dans un virus AAV dont la capacité de stockage est bien moindre : 4,7 kilobases environ.
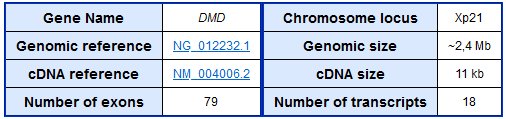
Source : e-dystrophin database. The gene.
Au moins 60% des mutations du gène DMD à l’origine de la myopathie de Duchenne et de celle de Becker sont des délétions plus ou moins importantes. Une délétion présente dans un allèle du gène entraîne la synthèse d’une dystrophine raccourcie. Il a donc semblé d'abord qu’une thérapie génique avec un allèle thérapeutique codant pour une dystrophine raccourcie ne pouvait être efficace. Des observations ultérieures résultant notamment du séquençage du gène DMD chez de nombreux patients ont conduit à nuancer fortement cette hypothèse.
C - Informations fournies par un cas de myopathie de Becker très peu sévère
Le phénotype clinque
Dans un article de Nature, S.B. England et son équipe de neurologues relatent les observations faites dans une famille où des membres sont atteints de la myopathie de Becker.
Un homme de 25 ans (numéro 1353 dans l’arbre) est venu en consultation suite à une histoire familiale où des hommes apparentés avaient été atteints de myopathie de Becker. Les neurologues ont étudié cette famille ce qui les a amenés à établir l’arbre généalogique suivant.
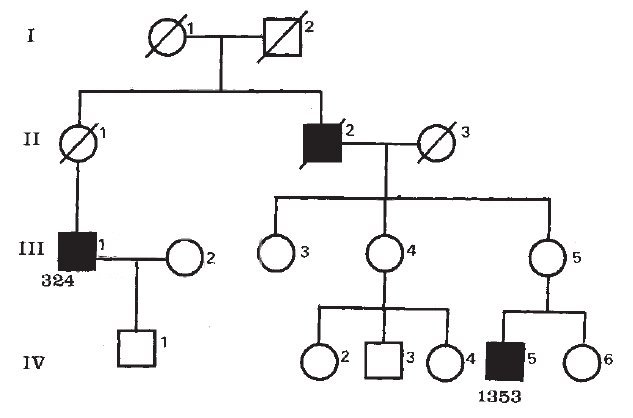
Source : Very mild muscular dystrophy associated with the deletion of 46% of dystrophin. Nature, 1990.
L’homme III1 (numéro 324) a manifesté les premiers signes de faiblesse musculaire à partir de la trentaine. Mais la progression de la maladie a été lente de sorte qu’à l’âge de 61 ans, il pouvait encore marcher à l’aide d’une canne, sans avoir nécessairement besoin d’un fauteuil roulant.
L’homme II2 est décédé d’une hémorragie, mais avant sa mort à l’âge de 52 ans, il avait montré des signes de faiblesse musculaire, tout en restant capable de se déplacer à l’aide d’une canne.
Les neurologues ont réalisé des biopsies de muscles de l’homme IV5 (numéro 1353) et constaté la présence de dystrophine normalement localisée dans les fibres musculaires mais plus courte qu’une dystrophine « saine ».
Les symptômes des hommes malades de cette famille confirment qu’ils sont atteints de la myopathie de Becker et on peut préciser d’une forme très légère comme l’indiquent les auteurs de l’article. Cela signifie que l’allèle du gène DMD qu’ils possèdent code pour une dystrophine au moins assez fonctionnelle.
Les caractéristiques de l’allèle muté cause de ce phénotype : « myopathie de Becker très peu sévère »
Les généticiens consultés par les neurologues ont séquence cet allèle. On dispose de la séquence codante de cet allèle ainsi que la séquence en acides aminés de la dystrophine qu’il code.
Fichier pour Anagène : Séquences DMD patient 1353.edi
La comparaison de la séquence codant de l’allèle muté du gène DMD possédé par les hommes malades avec celle de l’allèle de référence révèle que la mutation de l’allèle muté est une délétion dont on peut préciser les limites : elle débute au nucléotide 1993 et se termine au nucléotide 7008. Cela fait une délétion importante de 5015 nucléotides. On constate aussi qu’en amont et en aval de la délétion la séquence mutée est identique à celle de référence.
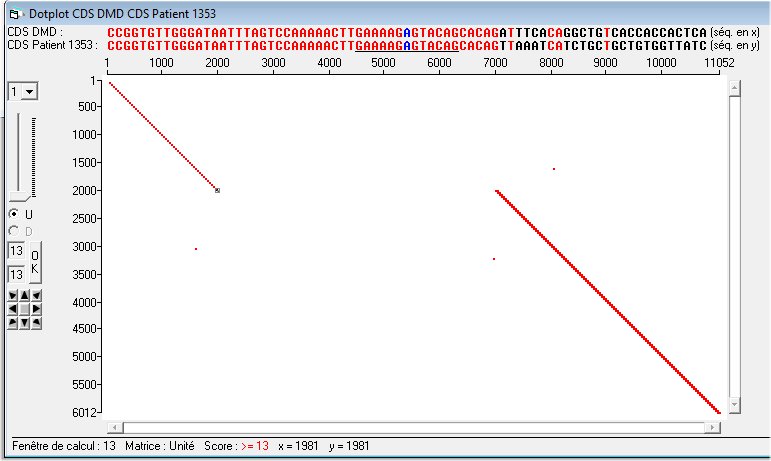
Mais c’est la comparaison des dystrophines qui est la plus informative. La comparaison globale de la protéine mutée et de la protéine de référence montre que la délétion mise en évidence au niveau nucléique se retrouve dans les séquences de la dystrophine. Le premier acide aminé délété est Gln en position 664 ; le dernier acide aminé délété est Glu en position 2335.La délétion est donc de 1672 acides aminés (2336-664). Comme la séquence de la dystrophine complète comporte 3685 acides aminés on calcule que la délétion présente dans l’allèle muté représente 45-46% de la dystrophine complète. C’est une délétion importante qui contraste avec la très faible sévérité de la myopathie des patients de cette famille.
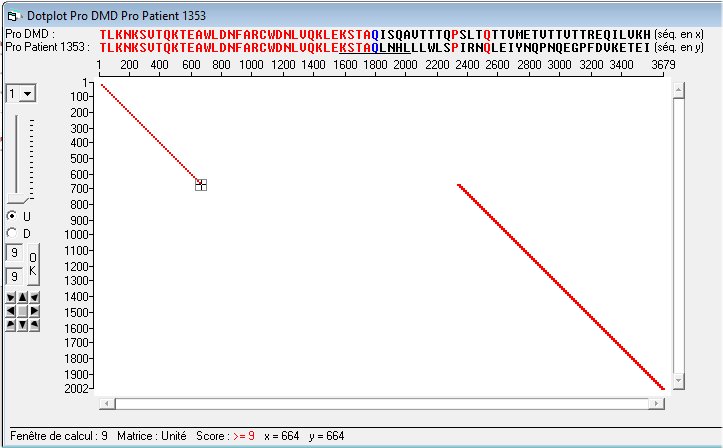
Cela indique qu’une dystrophine raccourcie peut conserver son efficacité, demeurer en grande partie fonctionnelle.
Les séquences de la dystrophine mutée en amont et en aval de la délétion sont identiques à celles de la dystrophine de référence. Les figures sur la structure de la dystrophine et sur celle du gène DMD (exons et domaines) indiquent que la séquence en amont code notamment pour le domaine de liaison avec l’actine du cytosquelette et la séquence en aval pour les domaines de liaison avec le complexe de protéines membranaires et cytoplasmiques. Grâce à la conservation de ces domaines, la liaison qui solidarise le cytosquelette avec la matrice extracellulaire est préservée.
Ce qui est délété dans la dystrophine mutée est une partie du domaine central « rod » (les exons 17 à 48). On arrive ainsi à la conclusion qu’un gène thérapeutique codant pour une dystrophine raccourcie dans son domaine central « rod » peut être efficace dans un protocole de thérapie génique.
D - Comparaison de l’effet des délétions dans les myopathies de Duchenne et de Becker
Ce sujet a déjà été abordé dans le point 5 du dossier sur la myopathie de Duchenne : « A la recherche d’une explication génétique des différences phénotypiques entre la myopathie de Duchenne et celle de Becker. »
Dans l’exemple étudié on compare l’effet sur la dystrophine d’une délétion de l’exon 19 cause de la maladie de Duchenne avec celui d’une délétion de l’exon 48 en cause dans une myopathie de Becker. Les explications des différences cliniques entre le phénotypes engendrés par des délétions d’un même gène peuvent se résumer ainsi :
- Dans le cas de la myopathie de Becker, la délétion n’entraîne pas de décalage du cadre de lecture au cours de la traduction. La séquence de la dystrophine en amont et en aval de la délétion n’est pas modifiée ; elle est seulement raccourcie. C’est le cas de l’exemple de la délétion des exons 17 à 48 des patients de la famille précédemment envisagée.
- Dans le cas de la myopathie de Duchenne, la délétion affecte le cadre de lecture. La séquence après la délétion est modifiée ; il apparaît assez rapidement un codon stop. Il en résulte que les domaines de la dystrophine assurant la liaison avec le complexe de protéines membranaires sont supprimés de sorte qu’elle n’est pas fonctionnelle.
En conclusion un gène codant pour une dystrophine fortement raccourcie peut être pris en charge par un vecteur viral AAV. La dystrophine qu’il code peut être au moins partiellement fonctionnelle à condition que ses domaines qui assurent la liaison entre l’actine du cytosquelette et le complexe de protéines membranaires soient préservés.